第二课 基因与免疫
免疫系统是一个复杂而高度发展的体系。它的任务是简单的:去搜索和杀死入侵者。如果一个人出生时带有严重的免疫缺陷系统,易发生病毒、细菌、霉箘、或寄生虫感染而死亡。在严重的复合免疫缺陷中,酶的缺少意味着毒素废物侵入免疫系统细胞内殺死免疫细胞,这样便摧毁免疫系统。免疫系统细胞的缺乏还是迪乔治综合症的基础,胸腺的不确当的发展,既起因於过量的免疫应答又起因於一种“自动免疫进攻”(autoimmune attack)。哮喘、家属性地中海热、和局限性回肠(肠炎病)全部起因於免系统的过量反应,而自动免疫多腺综合症和一部分糖尿病是由於免疫系统进攻了“自身”细胞和分子。当不能区分敌我时,免疫系统的一个重要作用是区分入侵细胞还是自身细胞应引起自动免疫疾病疫。
哮喘影响美国包括儿童在内的人群超过5%,它是以咳嗽为特征经空气传播的慢性炎症疾病。短促咳嗽胸部紧张。各种“确发”可以诱导疾病发生,或恶化哮喘发作。诱导“确发”因素包括病毒呼吸传染和受到如吸烟那样的刺激。哮喘的生理症状是由水肿引起的狭小空气通道(细胞组织空间流动),并从炎症细胞流入空气通道壁。
已知哮喘是一种“复杂”的遗传性疾病,这就意味着存在许多促使个人对疾病敏感的基因,染色体5、6、11、14全都和哮喘有关,有关这些基因在哮喘中的作用还不清楚,但根据研究,其中最基本的位置是在染色体5上。虽然哮喘在这位置上的基因还没有被特别确认,但已知这区域是应答哮喘炎症(包括细胞素、生长因素、生长因素受体)关键分子的基因编码区域。
特殊哮喘基因的探索正在进行,国际人类努力研究的得力助手是象鼠这样模型组织,鼠与人类一样,在它们染色体11、13、18上有同样的染色体5结构。对人类基因组这一区域基因的进一步研究将涉及到包括哮喘在内的特殊基因并能认识有关对哮喘发病机理起重要作用的生物学途径。

第二节 内分泌
内分泌系统是负责把激素释放至血液或淋巴中,内分泌系统缺陷由感染引起。梗塞或肿瘤破坏全部或大部分的腺体。然而,由于产生部分或全部破坏腺体结构的自动免疫反应,经常抑制免疫器官的活动。自动免疫疾病影响到一种器管后常继续損伤其它腺体,结果产生多腺体的疾病。
自动免疫多腺体综合症1型(APS1,也称APECED)是一种定位于染色体21的罕见自噬体隐性疾病。1997年末,研究人员报道了他们分离出一种新型的基因,他们称它为AIRE(自动免疫调节器)。数据搜索揭示出基因的蛋白产物是一种转录因子---一个在基因表达中起作用的蛋白质。研究人员显示这种基因突变归应于致癌基因APS1.
APS1基因缺陷的确认是朝向开发基因诊断疾病的第一步,基因及其功能的研究将进一步促进找到潜在的疾病治疗方法及增加我们对其它自动免疫疾病的总体机理的理解。
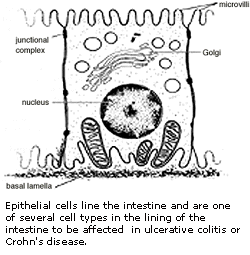
第三节 肠炎疾病
肠炎疾病(IBD)是在小肠和大肠引起炎症或溃疡的一组慢性炎症。IBD既可分类为溃疡性肠炎又可为局限性回肠炎。溃疡性结肠炎影响到结肠和直肠的内层,局限性回肠炎扩张至肠壁的更深层。由于是慢性炎症,所以一生中会多次复发。
大约有20%的局限性回肠性病有家属史。就是有这种复杂的踪迹,才意味着在基因组不同位置上的几个基因可能引起疾病。这种疾病的敏感位点最近被定位于16号染色体。在这些区域中找到的候选基因包刮与炎症应答相关的几种基因,包括与B-淋巴细胞功能有关的CD19;包括与白细胞粘结相关的sialophorin;包括与微生物细胞粘合相关的CD11整合素束;在IBDs中改变对IL-4调节功能感兴趣的白细胞介素-4。
因为某些与局限性回肠炎相关的遗传因子也影响溃疡性结肠,所以,对局限性回肠疾病的研究可进一步理解IBD的两种类型。
第四节 迪乔治综合症
迪乔治综合症(DIGEORGE SYNDROME)是一种罕见的先天性(例如,出身就有)疾病,个体之间临床表现差异很大,但最普遍的是包括经常发生感染史,心脏缺陷和面部结构特异。
迪乔治综合症是减数分裂(制造胚胎细胞和确认后代遗传差异的过程)重组发生差错,造成第22号染色体大量缺损所致。缺损意味着患有迪乔治综合症的病人在该区域中有几种基因不存在。出现疾病临床差异是与在染色体缺损中大量损失遗传物质有关。
虽然研究人员现在知道为正常发展胸腺和有关的腺体需要DGS基因,但扭转DGS的缺损是困难的。某些,例如心脏病患者和说话损伤者能通过外科或治疗获得效果,但免疫系统T—细胞(由腺体产生)的缺损是更具挑战性,并需对基因重组和免疫功能作进一步研究。

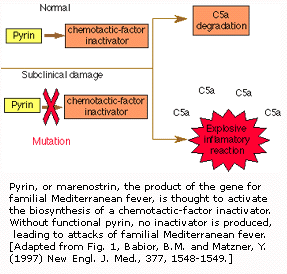
第五节 家属性地中海热
家属性地中海热(FMF),大多数发生在非-Ashkenazi Jewish,,Armenian,Arab和Turkish中,每200人中就有一个患这种病。每五个多点人群就有一个疾病携带者。FMF通常以周期性发热和腹膜炎(腹膜炎症)为临床特点的遗传性疾病。
在1997年,研究人员确认FMF基因并找到引起遗传性风湿病的几种不同基因突变。在染色体16上发现的基因给蛋白质作了编码,它几乎只有在对免疫应答起重要作用的白细胞上的粒细胞中发现。这种蛋白质可能在无“制动闸”作用的失活免疫应答控制下正常参与炎症产生过程。一种不适宜的全快速炎症反应就发生:FMF侵袭。
基因突变的发现将有助于为FMF开发一种简单的血液诊断试验。随着对突变蛋白的确认,有可能容易认识到引起疾病侵袭的环境触发因素,有可能不仅对FMF而且还可能对其它炎症疾病使用新的治疗方法。
第六节 具有过量-lgM的免疫缺陷
具有过量-lgM的免疫缺陷,是一种罕见的以增加许多有问题的lgM抗体而无力产生足够量的lgG和lgA为特证的原发性免疫缺陷。具有HIM的个体对周期性的细菌感染敏感并增加自动免疫和早年患癌症的危险性。
在对一种新抗原正常应答中,B细胞首先产生lgM抗体,随后B细胞打开产生lgG,lgA和lgE.就能产生更有效的保护组织和粘膜表面的抗体。来自HIM的最普遍现象是存在基因TNFSF5缺陷,TNFSF5基因在q26的染色体X上被发现。正常状态下,基因产生一种CD40的抗原配位子(CD154),一种在T细胞上对B细胞和其它免疫细胞上起到保证受体CD40作用的蛋白质。没有CD154,B细胞就不能接受从T细胞来的信号,就不能打开抗体产生lgA和lgG.免疫细胞之间缺乏CD40信号使得对HIM敏感的个体易受到如肺囊虫和隐孢子类的感染。
HIM的治疗主要由有规律的IV取代失去的lgG和对感染的及时治疗。然而,长久和最终的免疫只有骨髓移植才能保持,当找到合适的供体时就能做到这一点。

第七节 严重的组合免疫缺陷
严重的组合免疫缺陷(SCID)代表了一组罕见的以很少或没有免疫应答为特证的有时是致死的先天性疾病。以普遍熟知的“气泡男孩”病为代表的SCID特点是特殊的白细胞(B-和T-淋巴细胞)缺陷。白细胞保护我们免受外来病毒、细菌、和真菌的感染。没有功能性的免疫系统,SCID病人很易对如肺炎、脑膜炎、和水豆这类疾病重复感染,出生时就会死亡。虽然易受外界入侵,但采用如骨髓和干细胞移植这种新的治疗方法,可以救活80%的SCID病人。
所有形式的SIID,其半数是通过母系X-染色体伴性遗传,伴性的SCID起引于白细胞介素2受体伽码(IL2RG)基因的突变,正常情况下,伽码(IL2RG)基因产生常规的数个IL成分,IL2RG激活一个重要的信号分子JAK3。一旦位于染色体19上的JAK3引起突变,也能产生SCID,有缺陷的IL受体和IL受体通道在区分入侵者及激活、过度发展调节其它免疫细胞方面起重要作用的T-淋巴细胞的过度发展。
SCID的另一种形式是在染色体20上缺少基因编码腺苷脱氨基酶,这就意味着酶的底物累积在细胞中,受影响个体免疫系统的免疫淋巴组织细胞遭到严重损害或完全失去。所有形式的SIID,其半数是通过母系X-染色体伴性遗传,伴性的SCID起引于百细胞介素2受体伽码(IL2RG)基因的突变,正常情况下,伽码(IL2RG)基因产生常规的数个IL成分,IL2RG激活一个重要的信号分子JAK3。一旦位于染色体19上的JAK3引起突变,也能产生SCID。有缺陷的IL受体和IL受体通道防止在区分入侵者以及在激活、调节其它免疫细胞胞方面起重要作用的T-淋巴细胞过度发展。
SCID的另一种形式是在染色体20上缺少基因编码腺苷脱氨基酶,这就意味着酶的底物累积在细胞中,受影响个体免疫系统的免疫淋巴组织细胞遭到严重损害或完全失去。
在SCID中心对“SCID鼠”进行新治疗的探索中,其最重要的发展是培育包括ADA、JAK3和IL2RG这些缺乏的基因。可通过人类组分去重新认识损伤鼠的免疫系统。所以这些动物为生物医疗研究中研究正常或基因突变免疫系统提供了非常有用的模型。
