第四节 测定酶活性浓度的两大类方法
一、固定时间法(取样法)
前面已经提到,早期临床实验室测酶活性时常使用比色法,先让酶与底物在一定温度下作用一段固定的时间,然后停止酶反应,加入试剂进行化学反应呈色测出底物和产物的变化。由于比色法灵敏度较低,或由于手工操作不易控制好,常定在30分钟左右。历史上这类方法常被命名为“终点法”、“二点法”、“固定时间法”和“取样法”。大多数文献使用“固定时间法”。此命名指出了用这类方法测酶活性浓度,必须保证酶和底物在所选定的温度下作用时间要很精确,否则将引起较大误差。但“取样法”的命名更具有特征性。因它指出此类方法和下面将提到的另一类连续监测法相比较,最基本一点的是此类方法停止反应后才测底物或物的变化。很难进行连续监测,而另一类方法则不需停止酶反应,就可测定反应物的变化,很容易观察到反应的整个过程。后一类方法开始于50年代,Warburg首先用分光光度计。在340nm处直接监测到反应物NADH的动态变化过程。由于不停止酶反应,不需添加其它呈色试剂。测定方法简单,结果准确,逐步取代了“取样法”成为目前测酶活性的主要方法。
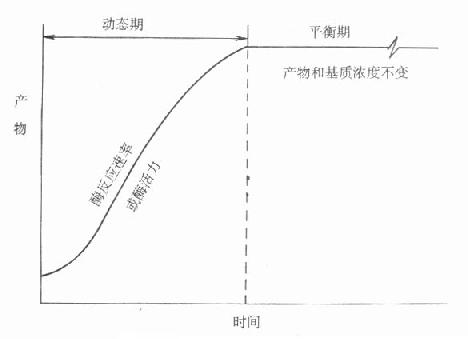
由于经历了一个发展过程,命名比较混乱而且大部分不甚确切,IFCC建议命名为“连续监测法”,不用目前广为流行的“动态法”术语。其原因是目前测酶活性浓度方法都是基于化学反应。图17-6是一个典型的化学反应过程。
图17-6 动态和平衡概念的示意图
可以看出整个反应过程,可以大致分为二个时期。一开始为动态期,化学反应按一定速度进行,反应物浓度随时间而变化,且变化程度不断变小,到一定时间。化学反应或者达到平衡,或者反应达到终点,此时反应速度为零,反应物浓度不变。最近IFCC文件按所用方法所测定的反应是在达到平衡前还是平衡后。分别命名为动态法或平衡法。一般文献往往将后一类方法称为终点法。
二、连续监测法与反应进程的分析
从这个权威性的方法分类来看,前面所提到的二类测酶活性浓度方法都应归纳为动态法。目前广为流行的“动态法”命名显然是不确切的或者是不合适的。应该更多采用IFCC文件中建议的“连续监测法”命名。
从酶反应曲线来比较此二类方法。图17-7是一个典型酶反应过程。

图17-7 酶促反应中基质[S]、产物[P]
和反应速率V在不同时间变化的模式图
在酶作用下,底物[S]浓度不断下降,随之有相应产物[P]产生。不少酶在反应一开始的阶段,由于各种因素影响,反应速度较慢,称为延滞期,随后在过量浓度的底物存在条件下,酶反应以恒定的速度进行,不受底物浓度变化的影响,这段反应称为线性反应期或零级反应期。随时间延长,反应速度除与酶量有关,还与底物浓度有关。即v=k(a-x),式中a为原来底物浓度,x为消耗的底物浓度,如反应物浓度项上指数为1,称为一级反应,假如反应速率受二种或二种以上物质浓度的影响,则各个反应物浓度项上指数总和作为反应级数,可以分别为一级、二级或多级反应,总的将这段反应时期称为非线性反应期。
很明显如测反应速度的目的是为了从此推算出酶含量的多少,则只有测线性反应期的反应速度才能作到此点。在延滞期、非线性期测的结果不准确,一般是偏低的。
取样法由于方法学上的限制,一般只测二管:一为没发生酶反应的对照管,另一管测酶作用一段时间后反应物的变化,实际上测得是平均速度,而且二管间隔时间都较长,在半小时左右,图17-8中t0,t分别代表不同时间二管反应物的量,虽然从t到t0反应物变化量是相同的。但实际反应过程是多种多样的。
只有当反应如曲线A时,用“固定时间法”才能测出真实的酶活性,实际工作中是很少见的,图中曲线B代表了最常见的反应情况,在一个很短的线性反应期后在大部分测定期间内主要为非线性反应期。曲线C说明在测定期间包括了延滞期,曲线D则不仅包括了延滞期,还包括非线性期,在这些反应中如用固定时间法来测定,结果是不够准确的,一般是偏低的,而且酶浓度愈高,偏离程度愈大。
假如从上节叙述中得出一个重要结论:即在设计和选择测定酶活性浓度方法时必须是在“最适条件”下测定最大反应速度。那么从这段叙述中可得出一个对上述结论一个很重要的补充,即所测的最大反应速度还应该是初速度即V0。
理论上的V0在实际工作中是不存在的,必须让酶和底物作用一段时间,消耗掉一定量的底物,才能测出反应速度,一般说,如消耗底物在5%内所测到的反应速度都可认为是初速度,如底物浓度很高时,底物消耗在20%以内的反应往往还在线性反应期。
连续监测法能满足上述重要要求。由于不需停止酶反应。很容易得到整个酶反应曲线,然后根据反应曲线情况,避开延滞期和非线性反应期,只在线性反应期测定最大反应初速度。同时由于分光光度法的高灵敏度,自动生化分析仪的高精密度,连续监测法能在很短时间,甚至在1分钟反应时间,准确测定反应速度,从而求出准确的酶活性浓度。“固定时间法”测定时间长达30分钟,很难满足上述方法学上测定初速度V0的要求。
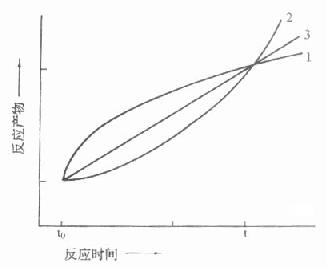
图17-8 二点测定法中可能引起的误差
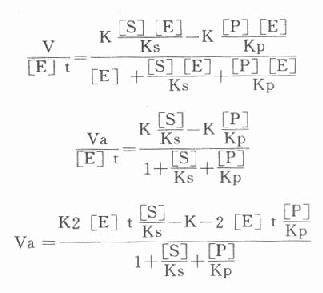
从理论上说,只有当测定的是初速度,米氏方程式才能成立,并可能测出真正的Km。用“固定时间法”测出的速度受到逆反应速度的影响。可以下式表示有逆向反应存在时的酶反应过程。
k1 k2 k3
E+S→ES→EP→E+P
← ← ←
K-1 k-2 k-3
存在着EP和P时,就会出现逆反应,此时所测的为一表观速度(Va)或反应的净速度。
Va=k2[ES]-k-2[EP]

[E]t代表酶总量,在反应过程中包含了游离酶[E]、酶底物复合物[ES]和酶产物复合物[]EP]。
Ks为[ES]的解离常数,Kp为[EP]的解离常数,故:

代入上式得:
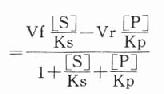
根据Maldanc公式Keq=VfKp/VrKs得:

在反应特定时间,产物[P]为一定值,上式得:
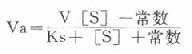
此公式从理论上说明,如所测反应有逆反应存在时,不存在原来的米-门方程式。用各种作图法很难求出准确的米氏常数。如果说所测酶反应不仅存在着逆反应,产物还抑制了酶反应,则动力学公式将更为复杂。这就是为什么不仅在建立方法时,就是在研究酶的动力学时,也总是希望所测量的都是酶反应的初速度。
以上这二类方法并不是概括了所有测酶活性浓度的方法,也并不排除在非线性反应期测酶活性浓度的可能性。如固定底物变化量,则不同标本达到此变化终点的时间,与酶量成反比例,Somogyi曾提出一种测淀粉酶的方法,酶与底物混合后。每隔半分钟取一滴出来加入碘液呈色,观察颜色由紫色变为红色时间,也就是淀粉酶全部水介变为糊精的时间,并由此时间的长短计算出酶含量高低,这类方法不需使用很高浓度的底物,有其独特之外,在自动生化分析仪中也曾使用类似的方法,值得人们重视。
三、连续监测法测酶活性浓度
连续监测法具有众多的优点,随着科学技术的发展,自动生化仪的使用,正在逐步取代“固定时间法”,发达国家从50年代到70年代末用了约20余年的时间,我国从80年代初开始使用连续监测法,虽然发展很快,但至少仍有不少地区医院实验室仍使用老的方法,就是已使用新法的实验室,也不一定掌握得很好,本节将比较详细地从分类,酶偶联反应,测定注意事项等方面对连续监测法加以介绍。
(一)连续监测法种类
⒈直接连续监测法这类方法是使用各种分析手段,如分光光度法、荧光法、pH计、旋光计、电导仪等等,在不停止酶反应条件下直接测反应体系中底物或产物的变化,从而计算出酶活性浓度,其中尤以分光光度法应用最多,应用最广的有NAD(P)H反应系统,可以测定大部分的脱氢酶,还有的是所谓“色素原”底物,其本身为无色或微黄色,在酶作用下生成有色化合物,适用于测定水解酶和一些转移酶。
⒉间接连续监测法直接法试剂简单,操作方便,可惜的是只有当底物与产物之间,在光学性质或其它理化性质有显著差异时,才有可能使用此法。到目前为止,大部分酶都无法用直接法测定。
科学家还曾设法在原来反应体系中添加一些试剂,这些试剂必须不与酶作用也不影响酶活性,同时又能与酶反应物迅速作用,产生可以被直接测定的物质,典型的例子是Ellman的胆碱酯酶测定法,底物为硫代乙酰胆碱,酶水解产物硫代胆碱与添加的二硫代硝基苯甲酸(DTNB)作用立刻生成黄色化合物,可在412nm用分光光度法连续监测。
实际工作中,更多的是在反应体系中加入另一酶试剂。进行适当的酶促反应,完全可以胜任上述工作。目前酶偶联法已经成为应用最广、最频繁测酶活性浓度的方法。
最简单的酶偶联反应为以下模式:
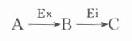
Ex为所要测定的酶,A、B二物质分别为其底物和产物,对此二物质变化,无法直接监测,此时可加入其它酶,其底物为Ex的产物B,其反应产物C可直接监测,这样通过第二个酶反应有可能推测出第一个酶的活性浓度,外加的第二个酶称之为指示酶Ei。
如果一些酶反应找不到合适的指示酶与其直接偶联,此时往往可以加入另一种酶,将二者连接起来,模式如下:

将这种联接的酶称之为辅助酶(Ea)。个别情况可能使用二种乃至二种以上的辅助酶。
酶偶联反应与一般酶反应的一个重要区别处在于其有一个明显的延滞期。酶偶联反应一开始只存在着底物A,不存在指示酶的反应,随着产物B的出现和增加,指示酶反应随之加快,Ex和Ei反应速度相等,也就是达到稳态。从酶反应开始至稳态期间,指示酶反应较慢且不稳定,称为延滞期。显然这期间指示酶反应速度不能代表测定酶量多少。设计或选择酶测定方法时,如果用酶偶联法,延滞期越短越好,测定时间一定要避开此期。
是不是所有类型的酶偶联反应都可用来测酶活性浓度?回答是否定的。因为测酶的活性浓度是依据测定酶反应速度――△A/△t或△B/△t求出。在酶偶联法,此值无法直接求出,而是通过测定指示酶反应△C/△t间接求出,要使酶偶联法测得的酶活性浓度准确可靠,则Vind=Vx。换言之,指示酶的最大反应速度必须等于或接近测定酶的最大反应速度。
当测定酶的反应速度明显大于指示酶,此时A很快转变为B,由于指示酶反应慢,中间产物B大量推积,最终产物产生速度明显慢于底物A的消失速度。
当指示酶速度加大后,中间产物B堆积减小,指示酶反应速率偏差程度也变小。只有当指示酶大量存在时,出现产物B就能将其迅速变为C。
可以看出在延滞期后(即B达到高峰后的时期),C和A的变化速度非常一致。也就是只有在些情况下,才是测定酶浓度的理想条件。
从理论上说,用酶偶联反应测酶活性浓度时,最好条件应是测定酶反应为限速反应。动力学上为零级反应,而指示酶为一级反应,酶反应速度与指示酶底物浓度相关。
(二)指示酶、辅助酶的种类和浓度
指示酶、辅助酶的种类:常规化验中常用的酶偶联法中,多以脱氢酶为指示酶,在常规化验中的自动分析仪几乎无一例外都有340nm波长,通过NAD(P)H系统可以很方便地监测到指示酶反应。但从理论上说,往往可以有不止一种偶联方法,只要设法使偶联反应中最后一个是指示酶反应,前面已提到测CK可以正向逆向二个方向建立二种不同酶偶联的反应。又如在丙氨酸转氨基酶(ALT)测定法中,正向反应后产生丙酮酸和谷氨酶,目前最常用的是用乳酸脱氢酶与丙酮酸偶联反应,伴有NADH下降。但也可以用谷氨酸脱氢酶与谷氨酸作用,伴有NADH生成。
总之,酶偶联法为实验室工作者开辟了一个广阔前景。在设计和选择测定酶活性浓度方法时,应该创造更新的方法,而不应拘泥于书本上的方法。
在选择时首先应考虑方法的特异性。在ALT方法,测定丙酮酸显然优于测谷氨酸,因为多种氨基转移酶都能产生谷氨酸,而只有ALT产生丙酮酸。还有干扰反应或副反应,在ALT测定中,由于正常以及病理血清中含有一定量丙酮酸,将受到它的干扰,又由于底物中含有大量α-酮戊二酸,可被血中谷氨酸脱氢酶作用消耗NADH。
其次应考虑酶偶联反应的合理性,如能直接与指示酶偶联,就没有必要加入辅助酶,所选用的指示酶、辅助酶除了应选Km小的酶(这样容易使它们催化反应速度大大超过测定酶反应速度),还必须考虑指示酶、辅助酶是在测定酶的“最适条件”下工作,此时往往不是指示酶、辅助酶的最适条件,如二者差异太大不利于整个酶系统的反应,例如乳酸脱氢酶最适pH为7.4,而丙氨酸氨基转移酶最适pH也是7.4,乳酸脱氢酶作为指示酶比谷氨酸脱氢酶更为合适,因后者最适pH为8.4。
如最后制成试剂盒,则还需从经济角度选择一些价廉物美,又易取材、纯化得到的酶制剂。
(三)指示酶、辅助酶的浓度
从上节描述中可以知道建立一个合适的酶偶联反应体系,不是很容易的指示酶,辅助酶的反应要能准确反映出测定酶含量,中间产物必须低,延滞期必须短,要作到此点,这些工具酶用量很大,但用量过大经济上不合算,所以在酶偶联测定法中,选用适当量的指示酶是一个重要的问题。一般可用反复试验法,即试验性地选用不同量的指示酸直到偶联酶反应中指示酶反应速度不随工具酶的增加而升高,并在所选用的“最适条件”下,指示酶反应速度和酶活性浓度成正比例。
这种方法工作量大,而且不一定能得到最适结果,较好的办法是可先从理论上进行计算,得出一个大约结果,然后在此范围内进行试验,这样可以节约时间和精力。
最简单的方法是根据Vx/(Km)x=Vi/(Km)i的比值来选择指示酶的用量Vi,式中Vx为测定酶的测定上限,(Km)x和(Km)i分别是测定酶和指示酶的米氏常数;有人计算过当比值为1:100时,测定值低4%;如改为1:10时,指示酶的反应速度将比测定酶反应速度慢28%,如增加到1:1000,则误差只有0.7%。
这是因为,当我们用酶偶联反应体系测酶活性浓度时,测定酶的反应必须是体系中的限速反应,工具酶所催化的最大反应速度必须远远大于测定酶,由于工具酶所催化的反应必须在中间产物浓度很低条件下进行,并且将其很快转变为最终产物,反应体系中不应有中间产物堆积,否则将导致误差。
下面举一实例,在肌酸激酶测定法中,CK的Km为2.4mmol/L,指示酶6-磷酸葡萄糖脱氢酶的Km为0.27mmol/L,如我们将CK测定上限定为450μ/L(实际测定时标本稀释60倍,实际为7.5μ/L),如希望上述比例为1:100。代入上式:
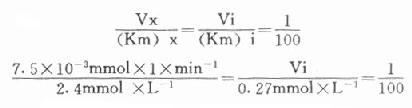
Vi=3.1×10-3×min-1×0.27mmol×1
=0.835×10-3×mmol×min-1
=0.835U
如反应体系为1ml,求出在此反应体系中只需0.835U的6-磷酸葡萄糖脱氢酶即可。
另一法可以根据米氏方程式来计算,在酶偶联反应中,指示酶催化速度(Vi)的米氏方程式为:
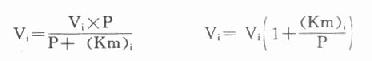
以天冬氨酸氨基转移酶(AST)为例希望中间产物P浓度很低,定为0.001mmol/L,指示酶苹果酸脱氢酶。Km为0.0165mmol/L,如设定AST上限为300U/L(标本为1:12稀释,实测上限为25U/L)。代入上式:
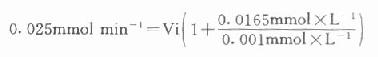
Vi=0.0014mmol min-1=1.4U
得1.4U,如反应体系为3ml,则试剂中苹果酸脱氢酶用量为466U/L,目前试剂中用量为600U/L。从以上计算来看,试剂中用量是足够的。
在酶偶联反应体系中,不可避免会出现延滞期,在测定酶时希望此延滞期愈短愈好,此时可利用Mcclure介绍的计算法计算出一定延滞期时所需指示酶的量。
Mcclure假定在下面酶偶联反应中:
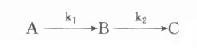
第一个反应为零级反应,第二个反应为一级反应,且中间产物B浓度远小于指示酶的Km,即B<<(Km)i。
通过推导,可得
dB/dt=k1-k2B
上式积分得:
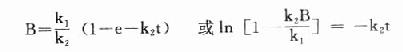
当t足够大时,e-k2t→0,此时B浓度不变达到稳态,此时B的浓度为Bss,代入上式:
首先可得 Bss=k1/k2
以及ln[1-B/Bss] =-k2t
同时在稳态下,k2是指示酶正向反应的常数,在一级反应中k2=Vi/(Km)i。
令F=B/Bss,此即在时间t时产物B为其稳态浓度的百分数,一般选用0.99。
最后可得公式:
 Vi=
Vi=
例如在CK测定中,指示酶6-磷酸葡萄糖脱氢酶Km为0.11mmol/L,我们希望延滞时间为1分钟,则指示酶用量为:
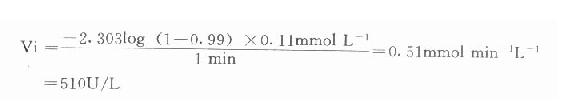
总之,从上式可清楚看到,延滞时间t与指示酶的Km成正比例,用量成反比。
四、酶活性的浓度单位
50年代以前酶活性浓度单位的命名混乱,常以方法提出者的姓氏来命名,例如淀粉酶的Somogyi单位,碱性磷酸酶的King单位等等,定义参差不齐,给临床医师带来很大不便,尤其在建立“连续监测法”测酶后,大量酶应用于临床,此混乱现象更为突出。1963年国际生化协会通过广泛讨论,提出一个国际单位定义来表示酶量的多少,即1分钟能转化1微摩尔底物(μmol min-1)的酶量为一个国际单位,以IU表示之,由于意见不一致,至今尚未指定酶反应温度,而同一量的酶,在不同温度时间为1分钟所转化底物的量将有明显差异。为避免临床上误认为只要是同一国际单位的酶量在国际上无论何处所测结果都应一致,目前大多数实验工作者常省略国际二字(简写也由IU改为U)。
临床上测定的不是酶的绝对量而是浓度,1963年并未明确规定用ml或L表示体积。旧的文献中可见到mU/ml,或U/L。目前在临床化学中,几乎都习惯用U/L来表示体液中酶活性浓度。我国由于近年来大量用自动分析仪和连续监测法测酶,已逐步不再使用各种古老单位,而使用U/L来表示酶活性浓度。
近年来国际上大力推广SI制,我国已明确SI制为法定计量单位制,SI制中酶活性单位为Katal,即1秒中转化1个摩尔底物(mol s-1)的酶量,Katal对体液中酶量而言显然过大,常用单位为ukatal或nkatal。
上述国际单位和katal间关系如下:
1U=1μmol min-1=16.67nmol s-1=16.67nkatal
在我国不论实验室还是临床医师对katal都不太熟悉,如报告使用katal/L报告酶结果时,最好同时注明相应的U/L。
(一)连续监测法中酶活性浓度的计算
前面已提到连续监测法优点之一是计算方便,不需作标准曲线或标准管,用分光光度计监测酶反应过程时,很容易求出反应体系每分钟吸光度变化,根据摩尔吸光系数可求出△A相当测定物质变化的微摩尔数,由于临床医师需要知道的是标本中而不是反应体系中酶的浓度,计算中要考虑标本的稀释倍数,假如比色杯光径不是1cm,则还应考虑光径不同对△A的影响,这样整个计算公式应为:
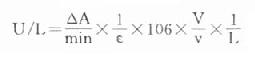
此中ε为摩尔(线性)吸光体系(mol-1・L・cm-1)
△A为吸光度变化
v为标本体积(ml)
V为反应体系体积(ml)
L为光径(cm)
在实际测定中后面几项皆为常数,所以上式常简化为:

(二)常数K的意义和设置
在测酶时,常数K值的选择是很重要的。此值过高虽然测定的线性范围较宽,但重复性差,反之,虽然精密度好,但线性窄。
此问题与仪器测定的嗓音(noise)密切相关,自动分析仪吸光度读数嗓音一般都需控制在0.001,也就是仪器须保证对同一溶液反复进行测定时,吸光度误差最好控制0.001上下,虽然此值不大,但已可能使测定结果产生1/1000K值的误差,如K值为6000,代入上式,则每分钟测定吸光度如有0.001微小变化,结果将是出现上下6U/L的误差,对于一此参考值较低的酶,如转氨酶而言显然太大,临床医师无法容忍这么大差异。
K值设置的首先出发点应是测定酶的判断值或参考值上限,应保证这些值测定的可靠,所以转氨酶的常数K一般在3000左右,不少人宁愿在1500左右,另外还应考虑到测定时间,一些半自动分析仪测定时间短到0.5分,此时0.001嗓音对每分钟△A误差将是0.002,反之,测定时间延长到2分钟,误差也小一半,也就是说,如测定时间长,则K值可以设置大一些,如测定时间只有0.5分钟,K值一般不超过4000。
改变K值最方便的途径就是改变标本稀释度,稀释倍数愈大,K值愈大。
(三)常数K值的检验
摩尔吸光系数(ε)对一定物质而言常是一个定值,但在一些外界条件影响下也会有所变化。最明显例子是对硝基酚,随pH不同,颜色差异明显,当pH>10时,405nm处其ε为18500,在pH7.0,ε下降为9400,颜色下降几乎一半。温度变化时,也会对NAD(P)H的ε产生一些影响。当波长大于334nm时,随温度升高,同一波长的ε值会轻度下降。
同时ε值是指用分光光度法,也就是在近似单色光的光源条件下才能成立,实际上大多数自动分析仪使用的是干涉滤片,波峰值可能出现差异,个别的半波宽可能为10nm乃至更大。由于杂散光的存在,会明显改变ε值,假如再考虑到注加系统的误差,实测K值很可能与理论K值不一致。Onuki检测了三台Hitachi-7150型自动分析仪测AST的K值分别为-5466、-5421、-5559。而理论K值为-6111则结果约增加为1.12倍。
测定实际的K值不是一件困难的事,目前有些自动分析仪已有相应的软件和试剂可以进行检测。事实上对一些性质稳定的物质如对硝基酚、对硝基苯胺,可以用高纯试剂配成标准品或直接购买可靠的校准品,将它们作为标本进行酶的测定,根据已知标准品的浓度和吸光度变化就可计算出实际K值,但此法不适用于测与NAD(P)H反应有关酶测定的K值。因为NAD(P)H不稳定,此时可使用测葡萄糖的紫外分光光度法试剂盒,对已知浓度的葡萄糖标准品进行终点法测定,此法产生与葡萄糖相等摩尔数的NAD(P)H,故不难从吸光度变化求出K值,也有人用乳酸脱氢酶测已知量的丙酮酸来求K值。
五、连续监测法中的干扰因素及其控制
大多数测定标本不是纯酶制剂,而是体液或组织液。其中除了测定酶外,还存在着其它酶和各种物质,如使用酶偶联反应,在反应体系中又外添了大量的各种酶制剂,因此在反应体系中可能出现一些我们不希望的副反应或旁路反应,这些都有可能对测定反应产生干扰。
(一)其它酶和物质的干扰
如组织匀浆中往往含有NADH-细胞色素C还原酶,它将干扰各种还原酶的测定。临床酶测定中最典型的例子是血液中丙酮酸对丙氨酸氨基转移酶测定的干扰,由于反应体系中含有大量乳酸脱氢酶和NADH,可与丙酮酸反应消耗NADH,引起340nm处吸光度下降,假如将此NADH下降也算为ALT活性将引起误差。其它如红细胞中腺苷酸激酶(AK)对CK测定的干扰,在CK的酶偶联体系中ADP是CK底物,但它又同时是AK的底物,二个酶反应都产生ATP,在工具酶(已糖激酶和6-磷酸葡萄糖脱氢酶)作用下都产生NADH引起340nm处吸光度上升,其结果是CK测定结果偏高,为避免此种干扰,所以在反应体系中加入AK的抑制剂AMP和二腺苷酸5′磷酸。
(二)工具酶的污染
目前试剂中所用的试剂酶多从动物组织或细菌中提取,不可避免地会污染有其它酶,如不注意此问题,会引起不正确结果,例如丙酮酸羧化酶催化下列反应:

可加入苹果酸脱氢酶和NADH进行测定,将草酰乙酸转变为苹果酸,并将NADH转变为NAP+。如果工具酶苹果酸脱氢酶不纯,含有乳酸脱氢酶,也可以作用底物之一的丙酮酸,同时消耗NADH,这样就无法准确测定丙酮酸羧化酶活性。
更有甚者,有些工具酶中就混有测定酶,这种情况下,将产生很高的本底。
所以所用的工具酶必须很纯,并检查污染酶的含量,例如测定天冬氨酸氨基转移酶所用的苹果酸脱氢酶中所含杂的AST时,按IFCC规定不应超过0.005%。
(三)非酶反应
有些底物不稳定,没有酶的作用就能自行反应,例如ALP的底物磷酸对硝基酚配成的底物溶液,室温放置过夜,即自行水解释放出对硝基成黄色。又如测醛缩酶时,其底物醛类化合物可以和NAD+起非酶反应产生一种具有类似NADH吸收光谱的化合物,给测定带来困难。
(四)分析容器的污染
如冲洗不当,分析容器和管道中混杂有各种物质,可能影响酶活性,如微量重金属可使酶失活,残留的表面活性剂可能抑制酶活性。
(五)沉淀形成
使用光学法监测酶反应时,如有沉淀形成或组织匀浆中颗粒的下沉都会引起吸光度变化,引起测定结果误差,此情况常见于底物溶解度低而反应体系中底物浓度偏高。可见于用γ-谷氨酰对硝基苯胺为底物测GGT时。
对上述的一些问题常可通过试剂空白管检出并加以校正。不同厂家的ALT,AST试剂盒由于杂酶存在,试剂空白管可以测出多少不等转氨酶活性,个别可达5U/L以上,这种试剂无法使用,较好的试剂盒也在2U/L左右。如不作试剂空白管对结果加以校正,所测结果将偏高,用半自动分析仪测定酶时特别要注意作试剂空白管,有时还需作标本对照管,例如测ALT时为除去GLD的干扰,可以在底物溶液中不加入丙氨酸,与标本中GLD作用引起NADH下降。
解决这些问题另一个有效措施就是不用单一试剂测酶活性,改用双试剂,先加的第一试剂常不含底物或底物之一,但含有所有其它成分,与标本作用一段时间待吸光度停止变化后,再加入底物开始测定酶的反应。
(杨振华)