
����ڡ�ijЩ�β�����������
����һ���Ҵ��ڸ��ڵĴ�л���Ҵ��Ը�����
�����Ҵ�����������˵��һ������ڳ���������ϸ���������������Ҵ����������ڣ�����Ի���Ӱ�첻�������������Ҵ���л��Ҫ��ָͨ�����ƶ��������ڵ���Դ���Ҵ������ڵĴ�л���Ҵ���θ��С���ϲ�Ѹ�ٱ����գ�θ30����С���ϲ�70����������ȡ���Ҵ�90��-98���ڸ��ڱ���л��ʣ�µ�2��-10����������������й���˵��Ҵ���л�ʴ�����Ѫ��Ũ�ȵ���ʧ������Ϊ100-200mg��kg��h��������ڽ�������ΪÿСʱ10g���ң�һ�մ�л��ԼΪ240g ��
�����Ҵ������������Ӳ���ķ�����֮�������й�ϵ��������Խ�ߵĹ��ң����Ӳ��������ҲԽ�ߡ�����������ÿ����10��/�����µ���䡢Ӣ��������ȹ��ң���Ӳ������������10/10�����£�����������ߵķ����˾�������27��/�����ң����Ӳ����������Ϊ30/10�����ϡ����ڳ������������γ��Ҵ���֬���Ρ��Ҵ��Ը��ס���Ӳ�����������и��������̥�����Ҵ��ۺ�����Ӱ��̥���ķ����ɳ�����ˣ��˽��Ҵ��������������л���Ҵ����л����Ӱ�죬�ں��ƾ���Ʒ���淺�ĵĽ������ɾ�����Ҫ���������塢�ٴ������������塣
�������Ҵ������ڵĴ�л
�����Ҵ������ڵĴ�л�����������������Ҵ�����Ϊҩ������ͬʱ��ÿ�����ͷ�7Kcal��1cal��4.2J�������ܣ��ڱ���ȡ���Ҵ��Ĵ֣�90��-98��������л�������ͷ���й�Ľ�ռһС��λ�����Ҵ��Ĵ��ڸ����ڱ����������Ҵ������л���ﲻ�������ڴ��棻�ݲ������ڵ����Ҵ������ٶȵ�����ķ������ơ�
�����Ҵ��Ĵ�л;�������Ҵ�����ø��alcohol dehydrogenase��ADH�������Ҵ�������ϵ����ADH�Ҵ�������ϵ���������Ҵ�������ϵ��microsomal ethanol oxidizing system��MEOS���������NADPH����ø-��������ø��ϵ�Լ�����������ø-��������ø��ϵ����10-14������Щ��ϵ����ADH�Ҵ�������ϵ�������Ҵ�������ϵ��Ϊ��Ҫ��
��10-14�������е��Ҵ���л��ϵ
������ADH�Ҵ�������ϵ����ȡ�����ڵ��Ҵ��ֱ���ϸ��Һ�е��Ҵ�����ø�������������ȩ����ȩ��һ������ȩ����ø����������������ᣬ�������γ�������øA����������øѭ����������ɶ�����̼��ˮ�����ͷ���������ATP��
������ȩ����ø�ɷ�Ϊ���ͣ�����ΪNAD�����Եĵ�Kmø��ȫ�ֲ����������ڣ�����Ϊ��Kmø���ֲ��������弰�����С��Ҵ�������������ȩ�����������ڱ�NAD�����Եĵ�Kmø�Ĵ�лϵͳ��������
�����������Ҵ�������ϵ��MEOS���Ҵ��Ĵ�л���ϸ������Ĺ����кܴ��ϵ������̬ѧ�Ϲ۲쵽���������Ƶ��˼�ʵ�鶯��ĸ�ϸ�������������������ӣ������Ҵ�Ҳ���������屻��л��ADH�Ҵ�������ϵ�������Ҵ�������ϵ����ɺ����ʲ�ͬ����10-15����
��10-15���Ҵ�����ø��ADH����ϵ�������Ҵ�������ϵ��MEOS���ıȽ�
| �� | ADH��ϵ | MEOS |
| ϸ���ڵ������Էֲ� | ��ϸ����Һ���������� | ��ϸ������ |
| ����pH | 10.8 | 7.2-7.4 |
| ��ø | NAD+�� | NADPH |
| ���ϳ�����Km�� | 2mmol/L | 8.6mmol/L |
| ��������Ƴ̶� | ����99�� | ����3�� |
| ��4.4mM/kg���أ� | �� | �� |
| Ͷ���Ҵ�������Ļ��Ա仯 | ���� | ���� |
| ���Ҵ��������������仯 | ���������ữ��ż�������⣬���� | ��NADPH��O2������ |
| ���Ҵ���л����ռ�ı��� | 75��-80�� | 20��-25�� |
����1970��Lieber��De Carli�ô����˸������MEOS����������̽�֣�����MEOS�ķ�Ӧ������������������pH����������Χ��pH6.8-7.4���ڣ��ڶԵ���Ҵ�����KmΪ8.2mmol/L������ҪNADPH��O2��Ϊ�������ӣ�����������ऺ�������Ϊ���ӹ���ʱ���ܷ�Ӧ���ܶ�CO��������У��ݶԹ�������ø��ADH����ø�����Ƽ����������ԣ��������ƶ�MEOSϵͳ�����Բ����յ���
�������Ҵ���л�Ի����Ӱ��
�����Ҵ���л�����������ĸ��ִ�л�쳣����������NADH/NAD+��ֵ������������ȼ���������
������NADH/NAD+��ֵ�������Ҵ������ڵĴ�л��һ�������Ҵ�������ȩ���ڶ���������ȩ���������ᡣ����������Ӧ���Ҵ�����ø����ȩ����ø�ĸ�ø����NAD+����ˣ����Ҵ������Ĺ�����NAD+����ԭΪNADH��ͨ���Ҵ���л�����Ĺ�ʣ��NADH�Ƴ�NADH/NAD+��ֵ��������NAD+/NADH��ֵ�Ľ��͡��������2.4g/kg�����Ҵ�����ʱ���ڸ��Ҵ���Сʱ��NAD+/NADH��ֵ��4.1����1.5��NADH/NAD+��ֵ����������NAD+/NADH��ֵ���½���ʹ�ø�������������ý��͡���һ���棬��ͪ�ᱻ�����NADH��ԭ�����ᣬ���������������ж����ֿ��������ж���������й������ϰ������·���������Ѫ֢��
��������ȩ�Ի����Ӱ�����Ҵ���л�Ի������Ӱ��������У����ܺ����Ҵ��ڸ��ڴ�л���м�����ȩ�����á���ȩ�Ļ�ѧ���ʻ��ã�����ǿ�ҵ�ҩ�����á�����ʵ���������ȡ�Ҵ����ܵĴ������ȩ�����������Ե��ڶ����飬������������Ҵ�����������Ĺ����ϰ����¡���������������ĸ�������Ĺ��ܲ�ȫ��������Ϊ��ȩ�Ķ������¡���ȩ������������Ĺ����ϰ���ʹ������ĺ������ܡ�֬�������������ܵ����ˡ������������߿����γ�һ�ֶ���ѭ�����������ƿ�������ڲ����϶���Ҵ�����Ũ�ȵ���ȩ�����ϸ������������ˣ��������������ȩ��л�ʽ��ͣ����¸�����ȩŨ�Ƚ�һ�������������幦�ܽ�һ�����¡�
������ȩ���˶����ڰ���л��������������ܡ��ļ������ʺϳ������������������⣬����������ҩ�����û������ã�����ȩ����ʹ��Դ�Զ���Ӱ��ͷŵĴ̼������������ã�������������Ҵ����ļ�����һ��ԭ����ȩ��������������ķ���������disulfiram��������ж����õ�ԭ����ȩ�����Ӱ����ϳ��������������ǰ�����ʽṹ�dz����Ƶ��������������һ�����Ǿ�����ԭ����ȩʹ5-��ɫ����л�����ϰ�������������лþ����õ�����-��-������������ƺ�����־����ϰ�������ȩ���Ҵ�����Ľ��֢״������һ��ԭ����ȩ�Ըκ��Եĸ�øA���Ծ�����ͬ�������ã�����ȩ���������������ά����B6ȱ��֢����Ҫ������ȩ����������Na+��K+-ATPø��L-����Ѫ�ᡢL-���װ���ȿɷ�ֹ��ȩ��ATPø���������á�
�������Ҵ���Ѫ�ǡ��������л��ˮ�����ƽ�⡢ά����D��л��ҩ���л��Ӱ�����ƺ�Ѫ���н������������ڽϳ�ʱ�䣨48-72Сʱ������״̬�¸����Ҵ��ܷ��������ĵ�Ѫ�ǡ������������NADH�����ߵ��±�ͪ��������-���ȡ�֬����������������ѭ���ȹ��̷����ϰ�������״̬����ȡ�Ҵ�ʱ��������������ϰ���������ȡ�Ҵ�ʱ��ʳ���㣬��ɸ���ԭ�������ͣ�����ǵ����Ҵ��Ե�Ѫ�ǵ�ԭ��
����ι�Ҵ������и���ƽ�������Ҵ��Ը�����ʱ�е����ʴ�л�ϰ�������Ͷ���Ҵ�������Ѫ�Ц�-�Ȱ���ת��ø�������ߡ��Ⱦ���Ѫ��Ȱ�������ø��������ø���������������������Ҵ��Ը�����ʱ����СҶ���IJ���ϸ�������巢�����ˣ���Ϊ�Ȱ�������ø��Ҫ�ֲ��ڸ�СҶ���IJ�λ��
�������ƺ�����������ǿ������ˮ֢״������ڿʡ��Ҵ����������Ǵ��忹�����ط��ڵ��������¡�Ѫ���и��ֵ��������ˮ��Ũ�������ƺ�����ѪҺpH���͡���̼���μ��٣������ж���������Ҫ�������Ҵ���лʱ��������ᡢ���ᡢͪ����������¡�
�����Ҵ��Ը����ˣ����Ҵ���֬���Ρ��Ҵ��Ը��ס��Ҵ��Ը�Ӳ����ʱ���ɹ۲쵽Ѫ����25-OH-ά����D�ļ��ͣ��併�͵�ԭ����Ҫ��ά������ȡ���㡢�Ҵ������ڳ�������������ϰ�������25-�ǻ����õĽ��ͼ����ڵĽ��͵ȡ�
�������������Ҵ��ж��������Ͱ���ҩʱ�ɼ������еķ�Ӧ����ʱ��������벻�����ض��ж�֢״����һ�������������߿ɶ����ʿ�ҩ�ԣ���������Ч������ѳ��֡��ڸ��ڴ�л���Ը��ж��Ե�CCl4���������������ȡ�Ҵ��Ķ�������ʾ��ǿ�Ķ��ԡ�������ʹ�������������ڶ����鲻�������ļ������������Ҵ�Ͷ������ɵ��¸�ϸ���������Ҵ���ҩ��ͬ�������б���л�����Ҵ���ҩ��ͬʱ��������ʱ�������ܷ��������乲ͬ����ϵͳϸ��ɫ��P450�ĵ���ľ���������������Ҵ�������ҩ���лø���Ե����ƣ��������Ҵ���ҩ�����ߵĴ�л������Ƶġ����ǻ�������ҩʱ������������ص��ж�֢״����ˣ�����Ӧ����������ҩʱ����ע����з������Ⱥá�
�����������Ҵ��Ը�������̥�����Ҵ��ۺ���
����������������������Ҫ���Ҵ��Ը����ˣ��Ҵ���֬���Ρ��Ҵ��Ը��ס��Ҵ��Ը�Ӳ���Լ�����ʱ�����и����ƶ����̥���쳣��̥�����Ҵ��ۺ�����fetal alcohol syndrome��FAS����
�������Ҵ���֬���ι��������Ҵ�����������֬���εı仯���Ҵ��ж���֬���εķ����ʿɴ�70��-80��������֬������Ҫ�Ǹ�������֬����������ɵġ�����֬������Դ����������ȡ��֬����������֯�е�֬���������ϳɵ�֬����
�����Ҵ���֬�����γɻ��ư������ټ����Ҵ��ж�ʱ֬���ζ�Ա�����ӣ�һ�δ��������Ҵ���ͨ������Ӱ�����������֬���Ķ�Ա��ͬʱ���и�֬Ѫ֢�ķ�������������ȡ�Ҵ�ʱ�Ҵ���л������NADH/NAD+��ֵ������ʹ������DZ�ͪ���-������͵�ת�����ӣ������ڸ��ڸ��������Ĵ����ϳɣ�������NADH�����ӣ�NAD+�ļ��٣�����������ѭ����֬��������������ƣ�����֬������������ͣ��ܴ�����ȡ�Ҵ�ʱ֬���ĺϳɼ����ڼ��١�
�����Ҵ���֬���������ԵĿ����ԵIJ�̬���������ƶ����ˣ�������Ϊ�Ҵ���֬���������Ҵ���л��������Ĵ�л������ɵĺ����
�������Ҵ��Ը����Ҵ��Ը����ڲ�����֯ѧ���Ը�ϸ������Ϊ��Ҫ�仯��������С���γɡ����ڴ��ָ�ϸ�������Ļ��ƿ��������������Ҵ��Ը�����ʱ�ĵ��������͵��ķ����ϰ����Ҵ��Ը�����ʱ��������֬�������ӣ��Ҹ�ϸ��Һ���е��ף����ס��������ȣ����������������������������ˣ��Ҵ�����ȩ�������������壬������������ˣ��������ϸ�������йأ����������Ҵ���MEOS��һ��л��ϵ��ƽʱռ�ܴ�л����1/4����1/2����������������ȩ�IJ������ӣ�Ҳ��������������ˣ����Ҵ�����������ʱ�������ɻ�ʹ֬�ʹ����������¸��л�ԭ�����ĵļ��ٺ�����֬�ʵ����ӣ����Ҵ�����Ĵ�л����״̬��ɺ����������ӡ����Ҵ��Ը�����ʱ�������IgA���ࡢ��ϸ��ճ���������͵����߹����쳣��
������̥�����Ҵ��ۺ���̥�����Ҵ��ۺ����������и�������ɵ�̥���쳣������һ�ְ��������ϰ���������ϵͳ�Ĺ����ϰ����ɳ���ǰ��ʼ�ķ����ϰ��Լ�������ò�ͻ���Ϊ�������ۺ�����FAS�Ķ���ģ�ͱ�����̥�����ء������������϶�����Ϊ�ͣ�C-���������Ժ˵��IJ�����٣�������RNA��tRNA���١������FAS�������ڵ����ʺϳɵ����ƿɽ�������FAS�����ܷ����ӳٵ������ڴ������Ʋ���Σ������Ľ�������Ӱ����һ���ķ����ɳ���
����������Ӳ��������
������Ӳ���벡���Ը��ס����Ծƾ��ж����ж��Ը����������й�ϵ���ڸ�Ӳ���Ļ������з����ΰ������ԡ�
������Ӳ��ʱ�ɷ����Ǵ�л�쳣�������ԭ�ļ��٣��������л�ϰ���ʹ�����ᡢ��ͪ�ἰ��-ͪ��������ࡣ֬���л�е��̴����ĺϳ��ϰ�����Ӳ�����ڵ��̴��ĺϳ�����ϰ����ڵ����ʴ�л���棬���ס���ά����ԭ�ȵĺϳɼ��٣��������л�����غϳ��쳣���������г��ְ������Ѫ�����ߡ�
�������⣬��Ӳ��ʱ�ε�����ת�����Լ������Ĺ�������ϰ����ڸ�Ӳ���γɹ����У���ά��֯�γ�����Ҫ����̬�仯����ȱ������֢�̼��£���ԭ��ά�ĺϳ���ǿ����������Ӳ��ʱ�����ڵ�����ճ�������ӣ��ر��Ǻ��а����ǰ�������������B�������ӣ����뽺ԭ��ά���йأ�����ά��ʱ���ӵĽ�ԭ�Ԣ��ͼ����ͽ�ԭΪ����
������ʮ�������ڸ���ά�����о��нϴ��չ���������������ά���йص����ӣ���ϸ���ɷֶ��ԣ��ݷ�ϸ����Kupffer cell��KC������֬ϸ����fat-storing cell��FSC���ڸ���ά���γɹ����ж����ŷdz���Ҫ�����ã�����άϸ������ž������Ľ����֯�γ��йء����⣬��ԭ���ر��Ǣ��͡����ͼ����ͣ������Ե����Լ�ϸ������ʣ����������Ľ�ϵ����ʼ�ճ���ǣ���ijЩ��ص�ø�����������ø����ԭø��������ø��������һЩ����ά���Ļ���ӣ����ϸ�����آ������������ӡ�ת����������-��TGF-�¡������ء�ǰ������2�ȣ��ȶ������ά���йء�
��������ά��ʱ�ݷ�ϸ���ɷ��ڶ���ϸ�����Ӽ���ԭø������������ʣ����ɴ˶Ը�ϸ������ʵĺϳ��뽵����������ã��Ӷ��ڸ���ά���ķ�����չ��������Ҫ���á�����Ϊ��֬ϸ���Ļ�Ǹ���ά���Ĺؼ�����֬ϸ��������ԭ��ϸ��������е��ǵ���-����أ�LN������ά���ӵ��ף�FN���ȵ�����ʹ�����ڸ����˺���֯������ά���ķ���������Ҫ���á�̽���⼸��ϸ����ϸ��������γɵ�Ӱ�죬������ԭ�ϳɵĵ��ڻ��ƣ������ڲ�������ά������Ӳ�����������ơ�
���������λ��Ե���������
�����λ����ֳƸ����Բ����������صĸβ����µ�������ϵͳ�������ң�����һϵ�о���֢״ֱ��������ԡ��λ����Ǹι��ܲ�ȫ����Σ�ϲ�֢������λ��ԵĴ�л���ؼ�Ϊ���ӡ���10-16�оٸλ���ʱѪҺ�ɷֵı仯�����Կ����λ���ʱ�Ĵ�л�쳣�Ƕ��ġ�
����Ŀǰ��Ϊ������Ĵ�л�ϰ������Ǹλ��Է��������ﻯѧ����������Ӱ��ж��������ʼ������ƽ���������������λ��Ե����ﻯѧ���ơ�
��10-16���λ���ʱѪҺ�ɷֱ仯����ӳ�Ĵ�л�쳣
| ��лָ�� | �仯 |
| ����������� | �� |
| ��Ѫ�� | ���� |
| ��ѪҺ�ӣ����� | ���� |
| ��Ѫ�������� | �� |
| �����㰱���� | ���� |
| ��֧�������� | ���� |
| ��Ѫ�尷�� | ���� |
| �ɺ����� | ���� |
| ��δ��ϵ����� | ���� |
| �˷�߲�� | ���� |
| ���Ǵ�л | �� |
| ��Ѫ�� | ���� |
| �Ʊ�ͪ�ᣬ���� | ���� |
| �Ǧ�-ͪ����� | ���� |
| ��֬��л | �� |
| ��Ѫ������֬���� | ���� |
| ��Ѫ�����֬���� | ���� |
| ������ʼ����ƽ�� | �� |
| ��Ѫ��Na+�� | ���� |
| ��Ѫ��K+�� | ���� |
| ��Ѫ��pH | ������� |
�����就�ж���λ���
����40����ǰ�����ָλ����백��л�����й�ϵ���������ǣ��ٶ����λ��Բ���Ѫ�����ߣ������תʱѪ�����ͣ������Ըβ���������ߵ�����ʳ���ҩ����շ��λ��ԣ��۸λ��Բ����Ե�ͼ�仯��Ѫ��ƽ�У��ܶ���ǻ�����Ǻ�Ȯ���Ըߵ�������Ѫ����������֢״���ݸ���ע�����泥���Ѫ����200-400��g/dlʱ����ʾ���˵ĸλ������Ƶ�֢״�����������˼�ʵ�鶯�ᆳ����Ѫ���Ʒ����鳣�ɺ�ת��������ʵ���������ж���λ����������й�ϵ��
�������ڰ��ж��Ļ��ƣ�����Ϊ�����ڸι��ܲ�ȫ����£�Ѫ������Դ�����ȥ·���٣�����Ѫ�����ߣ�����֯�����Լ�Ϊ���У���������Թ����ϰ������»��ԡ�����Ѫ�����Գ���������ÿ��Լ4g�������ڰ���������֯�����ȣ�����������ԵĻ�����Ҫ�����ڵ����غϳɡ����ڸ����ز��䵼�¸ι��ܲ�ȫ���������������Ϊ���ͣ���֮��ǻ������·��ʹ�ɳ��ܻ�ѪҺ�İ������νⶾ��ֱ�ӽ�����ѭ������ɸ߰�Ѫ֢��λ��ԡ���ʳ�����ࡢ��������Ѫ��������Ρ��Ÿ�ˮ�Լ�Ӧ��������Ⱦ�������Ѫ�������������ӣ��Ӷ����շ��λ��ԡ�
������������֯�Ķ����������ڰ���Ҫ�Ǹ������Ե�������л��ʹ�������ữ���ATP�ȣ�Ũ�Ƚ��͡�������ϸ����л�ĸ��������������棺�ٰ������Ʊ�ͪ������ø�Ļ��ԣ�Ӱ��������øA�����ɣ��ȸ�����������ѭ������ʼ���裬��Ӱ��������������������ɣ��ڰ��ж�ʱ���������γɹȰ������ķ�ʽ�ⶾ���Ӷ������˽϶��NADH����-ͪ����ᾭ��ԭ�����������ɹȰ��ᣩ��Ӱ���������������ữ���������У�����ATP���ɣ��۴��������-ͪ����������ɹȰ��ᣬ��ʹ������ѭ���еĦ�-ͪ�����Ľߣ������˹�����������ϸ�����������ͷ���ת�������ڦ�-ͪ����ἰ������������ͨ��Ѫ�����ϣ�����ת��ø���Եͣ�����ʹ��-ͪ�����ȵõ����䣬��˰��ж�ʹ��ϸ��������ѭ�������ϰ���ATP�����ɼ��٣��ܰ��Ȱ���ϳɹȰ�����ʱ����ATP���ģ��ݰ��ܼ�����ϸ��Ĥ�ϵ�Na+��K+-ATPø������K+�о������ã�Ӱ�����ӷֲ����������������С�
����Ӧ��ָ�������ж������ܽ������иλ��Եķ�������Щ����Ѫ�������ߣ���Ѫ���Ʒ��һ����Ч������̽���������ơ�
�����������ѧ˵
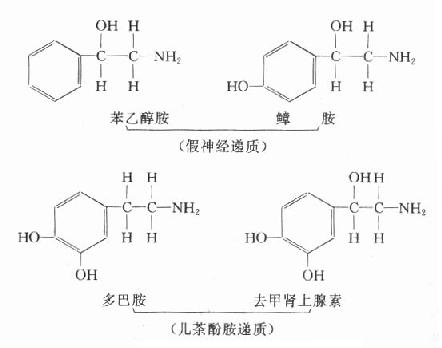
�����ڳ����ڣ�һ���ְ����ᾭ�����İ���������ø���ö��γɰ��࣬�籽�����ἰ�Ұ��������γɱ��Ұ����Ұ�����������¿ɱ����ڵ�������ø�ֽ��������ι��ܲ�ȫʱ�����ڸ��ڵ�������ø���Խ��ͻ������֧ѭ�����γɣ����Ƿ��㰷��ֱ�Ӿ���ѭ�����ԣ������ڷ������ǻ�ø���ã����DZ��Ұ��ǻ������ɱ��Ҵ������Ұ����ǻ��������J������-���Ұ������ڱ��Ҵ������J�������Ӱ����ʣ���Ͱ���ȥ���������أ��ṹ���ƣ��ֲ��������ش��ݳ嶯���ʳƼ����ʡ�
���������ʱ��ͷź�������ϵͳijЩ��λ�����Ը���״�ṹ���м���ϵͳ�����ܷ����ϰ���ʹ���Է���������ƶ����ԡ����ʡ���״��ͨ·�еĶ�Ͱ����ٵ���ȡ����ʹ�������������ռ���ƣ���������������������Ȼ���ٵ���ѧ˵Ҳ���ܽ���ȫ���λ��Եķ������ƣ�ͼ10-3����
�����簱���ƽ����λ���
������20�������ڸλ���ʱ�������л�쳣�����ϱ����������ظι������˺�����ǻ������·�������£���������ԭ���������ڰ������л�쳣����ͻ���ı�����Ѫ��֧�������ᣨ�Ӱ��ᡢ�����ᡢ�������ᣩŨ�����Խ��ͣ������就���ᣨ�������ᡢ�Ұ��ᡢɫ���ᣩ�������ߣ��Ӷ������Թ����ϰ���
�������ڷ����就������Ҫ�ڸ��ڷֽ⣬���ι��ܲ�ȫʱ�������就�����ڸ��ڴ�л�����ϰ��������Ѫ�е�Ũ�����ߣ�֧����������Ҫ�ڼ�����֯�д�л�����ڸι��ܲ�ȫʱ�ȵ��ص������ϰ����ڸ�ˮƽ���ȵ��ص������£�֧��������������뼡����֯���ֽ⣬���Ѫ���е�֧���������Ũ�Ƚ��͡�
���������就�����������֯������������ʵIJ������࣬ɫ�����ʹ5-��ɫ����������
ͼ10-3���ٵ���ѧ˵
�����࣬����Ϊ���������ʣ�ʹ�����������״̬����֧��������Ľ���ʹ��Ѫ�������Ϸ����就����ռ�����ƣ�ȱ���˾�������Ķ������Ǵ����������ڣ������������л�IJ�ƽ���������صĺ������������ѧ˵���ܽ��Ͳ�ͬ����¸λ��Է������Ƶ���Ҫ���档
�����λ��ԣ��ι��ܲ�ȫ���ļ����������У���Ѫ�嵨���ؿɳ������ĸ�ֵ���ܴ�40mg/dl����˵���е�֭��й�ϰ�����Ѫ������ͣ������䵰�ϳ������ͣ��۵͵��̴�Ѫ֢�����Ǻϳ������������£���AST��ALP�ɸ�ֵתΪ��ֵ�����ڴ�����ϸ���������������BUN�ʵ�ֵ����ʾ�κϳ����ع��ܵ��£���Ѫ�ǽ��ͣ����ڸ���ԭ�������٣�����Ѫøԭʱ���ӳ������ڸ�����Ѫøԭ�������ӡ������ӡ������ӵĺϳɼ��ͣ���Ѫ����ά����ԭ�ʵ�ֵ���ڸ��ںϳɼ��ͣ���Ѫ�����������غϳɽ������£���ѪҺpH���ߣ�PCO2���ͣ������Լ��ж���������ˮ������Ļ����������¡�
�����ġ���ʯ֢�����ﻯѧ
������ʯ֢����Ҫ��ɳɷ��е��̴��������ء��Ƽ�������Ԫ�ء���֭�ᡢ��ʯ���ʣ����ữ�ǵ����ǵ��ף��ȡ�����ʯ����Ҫ�ɷּ��γɻ��ƵIJ�ͬ���ɽ���ʯ��Ϊ���̴�ϵ��ʯ�������ؽ�ʯ��������ʯ�����࣬����ǰ�����൨ʯ���γɻ����������ܡ�
�����嵨�̴�ϵ��ʯ���γɻ���
�������̴�ϵ��ʯ���˵ĵ�֭���������쳣��
��������֭�е��̴��������ߺ��й����͵��̴��ĵ�֭�ǵ��̴��������Ⱦ���������֭�е��̴�����Ҫ���͵�֭���μ�����֬�ĺ�������һ���ı�����������̴���������֭���μ�����֬�ĺ������ͣ��ƻ�������֮����������������γ��ˡ���ʯ�Ե�֭�����б���ָ�������̴�ϵ��ʯ���˸��ڵ�HMGCoA��ԭø�����̴��ϳɵ�����ø���������ӣ���7��-�ǻ�ø����֭��ϳɵ�����ø�����Խ��ͣ������̴��ĺϳ����ӣ����̴���֭���ת�����٣������γɵ��̴������͵�֭�Ĵ�лԭ��
��������֭�е�֭���εļ��ٵ��̴�ϵ��ʯ���˵��ܵ�֭���л����С����Ϊ�����˵�һ�롣��һ�����������䵨�̴���л��ʧ������һ������������ڵ��ҶԵ�֭������������Ӽ�����ѭ���ϰ����¡����⣬����֬�����ȱ�����л�ϰ��ɵ��µ�֭�������ϰ���
��������֭�е�֭����ɵĸı������˵�֭�е��ᡢ���������ἰ�����������ߵı���Ϊ1.3��1.0��0.6�������̴���ʯ���˵ĵ�֭�ж���������ı��������Լ��͡�
��������֭�е���֬���͵��̴���ʯ���˵�֭�е���ֻ֬Ϊ������֭�е�1/3����֬/���̴���ֵ��������֭Ϊ6.6�����ڵ��̴���ʯ���˵ĵ�֭��Ϊ2.3����֭�е���֬��90������֬��������뵨֭���Ρ����̴���ͬ�γɻ���ŵ���Ҫ�ɷ֣���Ҳ�����ӵ��̴��ڵ�֭�е��ܽ��йء�
�������ĵ�λ�Ľ��� �ĵ�λ�ǻ��������������ɢ�����������ӷֲ����ȴ������ĵ�λ���λ��Խ�����Ŵ����Խ�࣬�ȶ���Խ��������֭�иʰ���֭����ţ�ǵ�֭��ı���ԼΪ3��1�����̴���ʯ���˵�֭��ţ�ǵ�֭����٣���������ɴ�15��1������Ե�֭��Ħĵ�λ��Ӱ�졣����ţ�ǵ�֭��������为��ɽ�ǿ�ģ�CH2SO3�������ӵ�֭�еĦĵ�λ��
�����ڸ������쳣��֭���ڵ������γ����ۿɼ��ĵ�ʯ���������ڽ�ʯ�γɹ����У���Ϊ��ʯ���ʵ��ǵ�����������Űѵ��̴��ᾧ������ճ����һ����������á�
�����浨����ϵ��ʯ���γɻ���
�����η��ڵĵ�֭�еĵ������ǽ���͵�������ȩ�ᵨ���ء���֭������֯�����Ħ�-����������ø���Խ��ͣ���ø����pH5���ң���֭pHΪ6.1-8.6���������ܸ�ø�������������Ƕ���-1��4-���������ƣ�����������֭�е�������ȩ�ᵨ���ز��ױ�ˮ����������õ��ܽ�״̬�������ظƽ�ʯ���˵ĵ�֭�пɳ��������쳣��
���������ڻ׳��������������˾�����ɵ�����Ⱦ����֭�г���ϸ���Ԧ�-����������ø������pH6.8-7.2���뵨֭pHһ�£��Ļ��Ըߣ������˵�֭�и�ø���������-�����Ƕ���-1��4-�����������Ƶ������������ʹ������ȩ�ᵨ���ش�����ˮ�⣬�������뵨���ض����ڳ�����
������������ϵ��ʯ���˵�֭�������Ƕ���-1��4-�����ĺ������������������鵨֭�и����ʵĺ���Ϊ200��g/ml���������ظƽ�ϲ��˵�֭�������40��g/ml����Ҳ���䵨�������ڳ�����ԭ����Ϊ��-����������ø������Ľ���������ϸ���Ԧ�-����������ø�������á�
�������ĵ�λ�������ǵ����ظ��ںϼ�����������ơ��ơ��ء�þ�������Ӽ�������Ⱦʱ���ֵĸ߷����л����ʣ�����ʹ�����ŵĦĵ�λ���͡�
�����ڵ����ؽ�ʯ�γɹ����л׳���塢�׳��Ѽ�������������ɽ�ʯ�ĺ��ģ�ϸ���Ԧ�-����������ø��������ȩ�ᵨ���ص�ˮ�⣬���뵨������Ca+�γɵ����ظƣ������ظ�����ǰ�������ӵ������£��ټ��Ͻ�ʯ���ʣ���Ҫ�����ữ�ǵ��ף����������ã�������ɵ����ظƽ�ʯ��
�����塢�ΰ�����������
������ijЩ�°�������ΰ������Ĺ�ϵ
���������������̣�AAF��AAF�ڸ��ھ�����ת���������AAF���뵰���ʷ����еĵ�����л�����������е������ʼ����ϣ���������߷��ӵĽṹ�빦�ܵ��쳣������DNA��ϣ�ʹϸ���ڵ��ص��ĺϳɲ��㣬ϸ��ʧȥ���ƣ���DNA���Ʋ���Ӱ�죬�Ӷ����𰩱䡣
����������ù���� ����ù����B1���������ں��ڸ�ϸ�������Ϲ�������ø����ת���ɻ���������ù����B1��������DNA��RNA��϶��°���
��������������ż������DAB�����ͻƣ����������ʵ���Ըΰ���DAB�ڸ�ϸ����Ϲ�������ø������N-���ǻ�����������ת��ø�������γ�N-SO3-O-������ż�����������������������ʼ����϶����𰩱䡣
������ΰ�ʱ�Ĵ�л�仯
�����������ʼ��������л�ı仯����֯�е����ʺϳ���ʢ������������֯�����ʵķֽ���ǿ���ΰ�ʱ���ΰ���֯���백����ֽ��л�йص�ø����ɫ��������ø���Ұ���ת��ø���հ�����ˮø����������ת���ἰ�鰱��ø�ȣ������������ͣ���������֯�а�����ֽ��л��������ʹ�������������ڵ����ʵĺϳɣ�����������방��֯�������йء����⣬�백����ת���йصĦ�-�Ȱ���ת��ø�����ڸΰ���֯���������ߡ�
�����ڸΰ���֯�������غϳ��йصĸ���֯����ø�����ᰱ������ת��ø��OCT��������������ϳ�øI��CPSI����������ø�ȵĻ��Խ��ͣ������ϳ��йص��춬���ᰱ������ת��ø��ATC������ϸ����ֳ����Ͱ��ϳ��йص���������ø��ODC���Ļ��������ߡ��ڸΰ���֯��֧��������ת��ø�Ļ��������ߣ�������빩���йء��ΰ���֯�а��ĺϳɽ��͡�������ʵ�������ڸΰ�ϸ��������ֳ�йص�ø�������ߣ����ϸ�������Թ����йص�ø���Խ��͡��ΰ���֯�������ϳɼ�̥���ף�����������֯�ķ��ֻ�������
�������ΰ�ʱ�Ǵ�л�ı仯�ΰ���֯�����Ǵ�л�йص�ø���Գ������仯�����������ؼ�ø������ϩ��ʽ��ͪ���ȼ�ø������-1��6-������ø��������-6-����ø�ȣ����Խ��ͣ��������Գ̶�Խ�ߣ���Щø����Խ�ͣ����ǽͽ�øϵ�����Ǽ�ø��������Ǽ�ø����ͪ�ἤø�ȣ��������ߣ��������Գ̶�Խ�ߣ���Щø�Ļ���Խ�ߣ���ͬ��ø�ı仯������̥�����ܱ����ڵĸ�Km��ͬ��ø�����Ǽ�ø��-���͵ȣ��Ļ����������ΰ���֯��ͬ��ø�ı仯��ʹATPʧȥ���ǽͽ�ĵ������ã�������ǰ�ϸ��ʧȥ��˹��ЧӦ��ԭ��֮һ���ΰ�ʱ�Ǵ�л�仯����Ҫ�ص��ǣ��ǵ������������ͣ���������������ռ99�����ͽ�ռ1�����ΰ�ʱ�ͽ��ռ50�������ǽͽ����ӣ����������٣���������;���Ĵ�л��ǿ����10-17����
�������ΰ�ʱ��֬���л�ڸΰ�ϸ���пɷ�����֬�ļ��ٺ������������ӡ���֬������з�֬�������ӣ�����֬�ʵ�֬�����г���C20��4�ļ��ٺ�C18��1�����ӡ����˵ĸΰ���֯���ܼ����������ԵIJ�����֬���ᣬ������������C20��39�����ֲ�����֬����ֻ��Ѫ��AFP���Եĸΰ����г��֡�
�����������ݱ���������ԭ���Ըΰ�����ֲ�Ըΰ�����ǰ�ڵȵĸ�ϸ����������ϸ����Na+��K+-ATPø���ԵIJⶨ����ǣ�ԭ���Ըΰ�����ֲ�Ըΰ�ϸ���и�ø�Ļ��Զ���������������ϸ������ǰ�ڸ�ϸ����Na+��K+-ATPø���������������ϸ�������ڰ�ϸ������ʾ�˸�ϸ������������Ʊû��Եı仯���ɡ��ΰ�ϸ��cAMP�ϳ�������Ϊ���ͣ��ΰ���֯�������ỷ��ø�������Ե�����������֯��
��10-17����������ΰ���֯�Ǵ�л�й�ø���ԵıȽ�
| �� | �� | �Ȼ��� | |
| ��л;�� | ø | �� | �� |
| �� | �� | ���������� | Ѹ�������ĸΰ���֯ |
| �� | ���Ǽ�ø | 100 | 500 |
| �ǽͽ� | ������Ǽ�ø | 100 | 229 |
| �� | ��ͪ�ἤø | 100 | 449 |
| �� | ������-6-����ø | 100 | ��1 |
| ������ | ����-1��6-������ø | 100 | ��1 |
| �� | ����ϩ��ʽ��ͪ���ȼ�ø | 100 | ��1 |
| �� | ��ͪ���Ȼ�ø | 100 | ��1 |
| ��������ͨ· | 6-��������������ø | 100 | 751 |
| �ǽͽ�/������ | ���Ǽ�ø/������-6-����ø | 100 | 8800 |
| �� | ������Ǽ�ø/����-1��6-������ø | 100 | 6463 |
�������ϸ������ԭ��
��������ϸ�������ԭ������ǰ����ѧ�°������ϸ����DNA������֮�⣬��10���������Ű��������ְ������������о��Ľ�չ�����Ǹ���ע�ظΰ��ķ�������ѧ���о���ϸ���ڱ������ڵ�ԭ����������������ѧ�������Ե��°����������±����ͨ����ͻ�䡢������λ�����������Ȼ��Ƽ��������ֹ��ȱ�����¸���İ������������ף��IJ��������ϸ���ڻ��������ص�ʧ�������յ���ϸ�����䡣����һ��������в�����ԭ������ı仯��ͬʱ�����ְ������ȱʧ��ʧ����Ұ��仹�Ƕ��һ�����ط������⼴��ΰ���ѧ˵����ν��ΰ���ѧ˵����˵����İ�������dz��ڶ�εط����ģ�����ϸ���ڰ������ְ�����ȵ��쳣�Ļ��۹����У����뿪��������ϸ����ֳ���ƻ��ƵĹ�����ֽε���ϸ��ת�����Ҷ��Գ̶������ӡ�
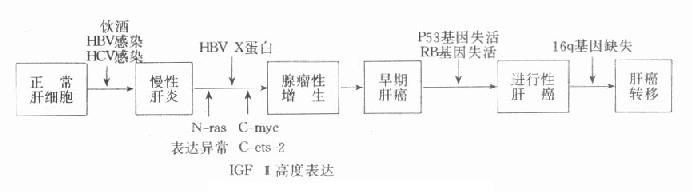
���������������ڸ��ף����ͼ����ͣ���ΰ��Ĺ�ϵ�����ܵ����ӡ������ײ�����HBV����ΰ��ķ�����ϵ�ѻ���������HBV�������е�X������ΰ������й�ϵ����ת����С��ʵ��֤����X������շ��ΰ��ķ�����X��������X����ʹ��ϸ����DNA�ϳ���ߵ���������״̬���ڶ�ΰ���������ٽ����á�X�����дٽ�DNA�ϳɵ����ã��ڸ�ϸ����������У�X����ĸ�ˮƽ�ij����Եı����DZ�Ҫ�ģ����ڰ���������������ְ�����ͬʱ���������Ƕ�ǿ����ϸ�����ķ��������Ƕ��ְ�����Эͬ���õĽ������N-ras�����쳣�ɵ��¿�Ĥ�źŴ��ݵĸı䣬����������ets-2��C-myc��P53�Ⱥ��ڱ�������쳣��ʹϸ���������ӣ�ϸ�����ڻ�Ծ��ֳ״̬��IGF-���fms������ǿ��ͨ�����Է��ڡ�����ʹϸ�����ڲ�������״̬�����������ۺ��������յ���ϸ����ֳʧ�ض��γɸ�ϸ���������ڸ�ϸ�����Ķ�ΰ���ѧ˵��ͼ10-4��ʾ��
���������ײ�����ΰ��ķ����������еĹ�ϵ��������Ⱦ��20�����ҵ��¸�Ӳ������30�����ҷ�չ�ɸΰ������ձ��ΰ���15�����������йأ���80����������йء�������PCR��������ϸ���������͡������ײ�������Ľ��������HBV-DNA������Ϊ69.5����HCV-RNA������Ϊ30.4�������ҹ����������ϸ�����Ĺ�ϵ�����������ܵ����ӡ�
ͼ10-4����ϸ�����Ķ�ΰ���ѧ˵