第四节 糖代谢的先天性异常
糖代谢障碍并不都出现血糖浓度异常,本节简要阐述糖代谢酶的先天性、遗传性缺损所致糖代谢异常。
一、糖原代谢先天性异常
最常见的是糖原贮积病(glycogenstoragediseases),这是由于糖原生成和分解的酶系统先天性缺陷所引起的一组糖原合成或分解异常、使糖原在细胞中过多贮积、或糖原分子异常的遗传性疾病。肝是糖原贮积病的主要受累器官,其次是心脏和肌肉(见表3-5)。
表3-5 各糖原贮积病及其临床体征
分型 |
酶的缺陷 |
受累器官 |
临床表现 |
| Ⅰ(von-Gierke病) | 葡萄糖-6-磷酸酶 | 肝、肾 | 肝明显肿大、发育受阻、严重低血糖、酮症、高尿酸血症伴有痛风性关节炎、高脂血症 |
| Ⅱ(Pompe病) | 1,4-α-D葡萄糖苷酶(溶酶体) | 肝、心、肌等 | 常常在2岁前心力、呼吸衰竭致死 |
| Ⅱ(Cori病) | 脱支酶 | 肌肉、肝 | 类似Ⅰ型,但程度较轻 |
| Ⅳ(Andersen病) | 分支酶 | 肝、脾 | 进行性肝硬化,常在2岁前因肝功能衰竭死亡 |
| Ⅴ(McArdle病) | 磷酸化酶 | 肌肉 | 由于疼痛,肌肉剧烈运动受限,否则病人可以正常发育 |
| Ⅵ(Hers病) | 磷酸化酶 | 肝脏 | 类似Ⅰ型,但程度较轻 |
| Ⅶ | 磷酸果糖激酶 | 肌肉 | 与Ⅴ型类似 |
| Ⅷ | 磷酸化酶激酶 | 肝脏 | 轻度肝肿大和轻度低血糖 |
| Ⅸ※ | 糖原合酶 | 肝 |
注:※糖原缺乏
由于酶缺陷不同,故糖原贮积病分为许多型。Ⅰ型糖原贮积病是1929年由VonGierke命名,所以也叫VonGierke病,为最常见的糖原贮积病。本型是因肝、小肠粘膜和肾脏的葡萄糖-6-磷酸酶或6-磷酸葡萄糖转位酶缺乏所致。前一种酶缺乏为Ⅰa型,后一种酶缺乏为Ⅰb型。该病为常染色体隐性遗传性疾病,发病率占人群比例为1/200000。
各型糖原贮积病的预后及治疗各不相同,故有必要对其进行鉴别(见表3-6)。在检测糖原贮积病时最可靠的方法是用肝或肌肉的活检标本测定特异酶活性。临床上有的病人还可见到一种以上缺陷的酶。
二、糖分解代谢途径的先天异常
糖分解代谢途径先天代谢异常可有丙酮酸激酶缺乏病,丙酮酸脱氢酶缺乏症和磷酸果糖代谢异常所致恶性发烧。
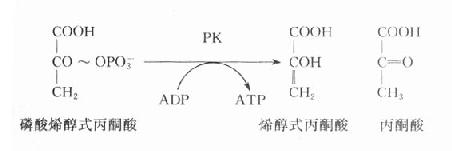
(一)丙酮酸激酶(PK)缺乏病
在糖酵解过程中,丙酮酸激酶催化磷酸烯醇式丙酮酸生成烯醇式丙酮酸,同时产生ATP,是酵解途径产生ATP的反应之一,PK缺乏将导致成熟红细胞缺乏ATP,进而发生溶血。
表3-6 各型糖原贮积病的特征
| 类型 | Ⅰ | Ⅱ | Ⅲ | Ⅳ | Ⅴ | Ⅵ | Ⅶ | Ⅷ | Ⅸa |
Ⅸb |
Ⅹ |
0 | |
| 受害器官 | 肝肾 | 所有组积 | 肝、肌、心 | 肝单核吞噬细包系统 | 肌肉 | 肝 | 肌肉 | 脑 | 肝 | 肝 | 肝、肌肉 | 肝 | |
| 糖原结构 | N | N | 短枝异常 | 长枝异常 | N | N | N | 巨大颗粒 | N | N | |||
| 酶缺陷 | G-6-P酶 | α葡萄糖 苷酶 | 脱枝酶 | 分枝酶 | 肌磷酸 化酶 |
肝磷酸 化酶 |
磷酸果粒糠酶 | 肝磷酸化酶 | 同左 | 糖原合成酶 | |||
| 空腹低血糖 | ++ | 0 | + | 0 | 0 | ± | 0 | 0 | ± | 0 | ++ | ||
| 对胰高血 糖素反应 |
血糖 | 0 | ↑N | 0↑ | ↑ | ↑ | 0± | ↑ | ± | N进餐0空腹 | |||
| 血乳酸 | ↑↑ | ↑ | ↑ | ||||||||||
| 半乳糖 果糖试验 |
血糖 | 0 | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ||||
| 血乳酸 | ↑↑ | ↑ | ↑ | ||||||||||
| 剧烈运动后血乳酸 | ↑ | ↑ | 0 | 0 | ↑ | ↑ | ↑ | ||||||
| 脂质代谢 | ↑↑ | N | 饿后 FFa ↑ |
|
运动后肌摄取 FA↑ |
TG↑ Ch↑
|
|
TG↑ Ch↑ |
TG↑ Ch↑ |
|
|||
| 其它诊断性试验 | 肝活检 | 红细胞抗原 ↑ | 红细胞糖原结构异常 | 红细胞磷酸化酶↓ | |||||||||
| 临床特征 | 生长停滞,肝大 | 心衰 | 似Ⅰ型但不明显 | 肝硬化腹水 | 肌无力 | 肝大 | 肌无力 | 意识障碍 | 肝大,常染色体隐性遗传 | 肝大伴性遗传 | 肝大 | 轻度肝大 | |
注:N-正常;O-阴性;↑-升高;↓降低;±-可疑
网织红细胞中含有线粒体,故可通过糖有氧氧化产生足量的ATP。而成熟红细胞中不含线粒体,完全依赖糖酵解供能。红细胞内生成的ATP主要用于维持细胞内外的离子梯度,特别是通过Na+-K+-ATP酶维持细胞内外Na+、K+浓度梯度。这对于维持红细胞双凹形状十分重要。若缺乏ATP,红细胞将发生肿胀,易发生溶血,实验室检查可以见到自身溶血试验阳性。PK的遗传缺陷是糖酵解途径中遗传性缺陷导致溶血性贫血的最多见原因。PK缺陷时,细胞中PK活性仅为正常细胞的5%-25%,故虽然PK缺陷少见,但其造成的溶血性贫血却对机体危害甚大。
(二)丙酮酸脱氢酶复合物缺乏症
丙酮酸脱氢酶复合物由丙铜酸脱氢酶、二氢硫辛酸转乙酰基酶、二氢硫辛酸脱氢酶及NAD+、FAD、CoASH、焦磷酸硫胺素、硫辛酸三个酶、五个辅助因子组成,其氧化的丙酮酸氧化脱羧生成乙酰辅酶A的反应是糖进入三羧酸循环、彻底氧化成CO2和水、产生大量ATP的关键。
在儿童中发现有多种丙酮酸代谢异常的疾病,其中有些是由于丙酮酸脱氢酶复合物中某些组份先天性缺陷所致。该酶复合物中各种亚基(催化亚基和调节亚基)都可能发生先天性缺陷。这些缺陷都可使丙酮酸不能继续氧化产生ATP,使脑组织不能有效地利用葡萄糖供能,进而影响了儿童大脑的发育和功能,严重者可导致死亡。
丙酮酸不能进一步氧化,致使患儿血液中乳酸、丙酮酸和丙氨酸的浓度显著升高,出现慢性乳酸酸中毒。丙酮酸脱氢酶的缺陷可以通过皮肤成纤维细胞培养并进行酶学测定予以测定。此类病人在一定程度上可通过进食生酮食物和限制糖的摄入使病情缓解或得到控制。
(三)磷酸果糖代谢异常
磷酸果糖激酶与果糖-1,6-二磷酸酶是作用相反的一对酶,它们所催化的化学反应是糖代谢途径中的一处无效循环(又称底物循环)。

由于酶的遗传性缺陷,以上无效循环得不到控制,造成ATP大量分解产热:ATP+H2O→ADP+Pi+热。临床上可因服用氟烷而诱发恶性发烧。
恶性发烧是一种罕见的遗传缺陷性疾病,其发病率占儿童的1/15000,成人的1/50000-1/100000。病人常因服用某种药物,如吸入氟烷而在几分钟内突然发病,表现为体温骤然升高、代谢性和呼吸性酸中毒,以及高血钾症和肌肉强直。人们认为,氟烷可以促进肌肉中上述两个酶所催化的耗能无效循环,诱发恶性发烧的产生。
三、其它糖代谢异常
(一)红细胞中6-磷酸葡萄糖脱氢酶遗传缺陷或变异
6-磷酸葡萄糖脱氢酶(G-6-PD)催化6-磷酸葡萄糖脱氢生成6-磷酸葡萄内脂,脱下的氢由NADP接受。此步反应是磷酸戊糖途径的关键部位,所产生的NADPH在维持红细胞的正常形态与功能方面起了重要作用。
G-6-PD缺乏病是伴性遗传性疾病,临床并不罕见。女性杂合子含有两族红细胞:一族酶活性正常,另一族缺乏G-6-PD。轻型G-6-PD缺乏病人红细胞中G-6-PD活性比正常人低10倍。在一般情况下磷酸戊糖途径提供的NADPH还能维持还原型谷胱甘肽的水平,保证红细胞的正常形态与功能。当红细胞中NADPH的需要量增加,如服用抗疟疾药扑疟喹啉时,正常人不会有什么危害,而G-6-PD缺乏病人红细胞中磷酸戊糖途径的代谢速度则不能相应增加,提供的NADPH不能保证维持还原型谷胱甘肽所应有的水平,可引起严重的溶血性贫血,俗称蚕豆黄。
(二)先天性家族性非溶血性黄疸
人类先天性家族性非溶血性黄疸(Grigler-Najjar综合征)是由于缺乏UDP-葡萄糖醛酸基转移酶,使胆红素不能与葡萄糖醛酸结合,形成结合胆红素,使胆红素以不易运输和排泄的游离形在体内堆积所致的先天性疾病。
正常人通过糖醛酸途径产生葡萄糖醛酸,后者在UDP-葡萄糖醛酸基转移酶的催化下,可与内源性如类固醇、胆红素和外源性如药物等物质结合,生成相应的葡萄糖苷酸化合物。结合型的葡萄糖苷酸化合物具有较强的酸性,在生理pH下有较高的溶解度,易于运输排泄。这在体内类固醇激素的灭活和胆红素的代谢,以及许多生物转化作用中具有重要意义。而病人因先天缺乏此酶,使其不但对胆红素代谢造成异常,同时也缺乏结合外源性物质生成葡萄糖苷酸化合物的能力。
(三)果糖代谢异常
果糖是食物中糖的一部分,主要来自蔗糖。从肠道吸收的果糖大部分在肝内通过1-磷酸果糖(F-1-P)途径代谢。代谢途径见图3-9。果糖代谢障碍是因与果糖代谢有关的酶缺乏所致。
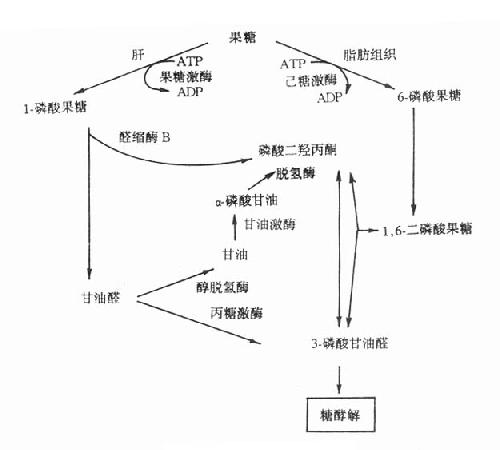
图3-9 果糖代谢途径
1.实质性果糖尿(essentialfructosuria)又称为原发性果糖尿症,它是由于果糖激酶缺乏所引起的常染色体隐性遗传疾病。正常人血中果糖比葡萄糖代谢快,其半寿期果糖为20分钟,葡萄糖为45分钟一次服用50g果糖后,通常在2小时之内血中果糖浓度就降至空腹水平(0-0.44mmol/L)。果糖激酶缺乏者一次服用50g果糖后,病人血中果糖浓度异常高,2小时内果糖仍未消失,并出现果糖尿。此型果糖尿又称为Ⅰ型果糖尿。病人无低血糖表现,这是因为病人机体内葡萄糖与乳糖代谢均正常。
2.果糖不耐受(fructoseintolerance)此病为常染色体隐性遗传性疾病,杂合子无症状。多数病人在断奶后给予蔗糖饮食时才发病,严重病例可致死亡。有些病例可由于手术前后给予果糖或静脉注射山梨醇引起严重肝、肾损伤时才发现。
果糖不耐受症的临术表现可明显不同。在婴儿可表现为呕吐、进食少、肝大、精神淡漠、生长停滞等。病人尿分析有果糖、葡萄糖等还原物质,多数病人有蛋白尿、非特异的氨基酸尿以及血浆转酶活性增高。有的病儿伴有其它肝、肾损害指标。在大龄儿童或成人则无症状,只在进甜食后,可有腹部不适、呕吐或腹泻。果糖不耐受的一个重要特征是服用果糖后出现严重的低血糖,病人即使肝糖原储备丰富也会在此时发生低血糖,这是由于过量的1-磷酸果糖抑制了肝磷酸化酶所致。
此症是由于1-磷酸果糖醛缩酶(醛缩酶B)缺陷引起,病人肝内1-磷酸果糖醛缩酶活性几乎完全缺失,而1,6-二磷酸果糖醛缩酶活性降低50%以上,造成肝内1-磷酸果糖的堆积及Pi和ATP的消耗。由于Pi大量消耗,肝线粒体氧化磷酸化减少,造成ATP缺乏。后者缺乏使肝细胞ATP依赖性离子泵功能障碍,膜内外离子梯度不能维持,细胞肿胀,细胞内容物外溢。

3.1,6-二磷酸果糖酶缺乏症此症为常染色体隐性遗传病,多在婴儿时发病。病儿表现为肌无力、呕吐、嗜睡、生长停滞和肝肿大等,感染可促使急性发作。若不治疗,在婴儿期就可死亡。
实验室检查可见空腹血糖低,即空腹性低血糖、酮血症、乳酸血症和血浆丙氨酸水平增高。诊断依据低血糖症,确诊需用肝、肾、肠活检标本测定该酶活性。
治疗主要通过食物疗法,食含果糖少的食物,少吃多餐,避免饥饿,一般疗效还可以,预后尚可。
5.半乳糖血症半乳糖在体内的正常代谢途径如图3-10所示。
影响半乳糖利用的各种因素均可引起半乳糖血症(galactosemia),但遗传性半乳糖血症主要有两种。
1-磷酸半乳糖尿苷转移酶所致的半乳糖症是最多见的遗传性半乳糖血症,其为常染色体隐性遗传疾病。患者半乳糖代谢终止于1-磷酸半乳糖阶段。杂合子病人此酶仅活性低下,但如果不持续服用半乳糖饮食,酶活性还可以维持健康。而纯合子病人此酶完全缺乏,如食用富含半乳糖的食物,病人会出现严重病变,甚至死亡。如果及时停用含半乳糖的食物,病人除智力障碍外,其他的各种症状均可消失。此型半乳糖血症临床表现为生长停滞,喂奶后呕吐和腹泻,继而出现黄疸、溶血、肝大、智力障碍,检查中可见患儿血清半乳糖水平明显升高,尿中出现半乳糖,红细胞中1-磷酸半乳糖尿苷酰转移酶缺乏。患儿进食半乳糖或乳糖后,常伴有低血糖和高半乳糖血症。
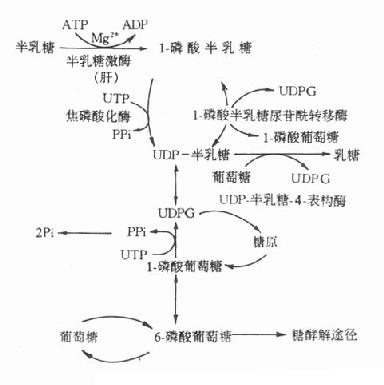
图3-10 半乳糖代谢途径
另一种遗传性半乳糖血症是由于半乳糖激酶缺乏所致,此型症状较轻,新生儿期不表现症状,往往发生白内障后才被确诊。
半乳糖血症的危害不是由于缺乏某种必需物质,而是由于产生毒性物质所致。半乳糖还原产物-半乳糖醇,在细胞内高浓度,如贮积于晶体,将吸收水进入晶体,造成晶体肿胀、混浊,引起白内障。此外1-磷酸半乳糖可能是某些毒性物质的前体,或者高浓度时本身就具有毒性作用。体外实验已证实,1-磷酸半乳糖可抑制磷酸葡萄糖变位酶和G-6-P酶的活性。
(四)粘多糖沉积症
因蛋白聚糖降解酶先天性缺陷所引起的蛋白聚糖分解代谢障碍,将导致产生各种类型的粘多糖沉积症(mucopo-lysaccharidoses)。其特征是过多的寡聚糖堆积与排泄。由表3-7可知,各型粘多糖沉积症的代谢基础相似,但遗传类型和临床表现各不相同。
表3-7 粘多糖沉积症的酶缺陷
| 名称 | 代号 | 酶缺陷 | 酶学测定样品 | 生化改变 | 遗传特性 |
| Hurler,Scheie综合征 | MPSⅠ | α-L-艾杜糖酸苷酶 | 成纤维细胞、白细胞、组织、羊水细胞 | 尿和组织中DS、HS增多,成纤维细胞中DS增加 | 常染色体隐 性 |
| Hunter综合征 | MPSⅡ | 艾杜糖醛酸硫酯酶 | 血清、成纤维细胞、白细胞、组织、羊水、羊水细胞 | 同上 | X连隐性 |
| Sanfilippo综合征A | MPSⅢA | HS-N-硫酸酯酶(硫酰胺酶) | 成纤维细胞、白细胞、组织、羊水细胞 | HS在尿中和组织中增多、DS在成纤维细胞中增多 | 常染色体隐性 |
| Sanfilippo综合征B | MPSⅢB | α-N乙酰葡萄胺苷 | 血清、成纤维细胞、白细胞、组织、羊水细胞 | HS出现于尿中 | 同上 |
| Sanfilippo综合征C | MPSⅢC | 乙酰基转移酶 | 成纤维细胞 | HS出现于尿中 | 同上 |
| Morquio综合征 | MPSⅣ | N-乙酰半乳糖胺-6-硫酸酯酶 | 成纤维细胞 | KS和CS出现于尿中 | 同上 |
| Morquio综合征 | β-半乳糖甘酶 | 成纤维细胞 | KS出现于尿中 | ||
| Maroteaux-Lamy综合征 | MPSⅥ | N-乙酰半乳糖胺4-硫酸酯酶(芳香硫酸酯酶B) | 成纤维细胞、白细胞、组织、羊水细胞 | DS出现于尿中 | 同上 |
| β-葡糖醛酸苷酶缺乏症无名疾病 | MPSⅦ | β-葡糖醛酸苷酶 | 血清、成纤维细胞、白细胞、羊水细胞 | DS、HS(±)出现于尿中 | 同上 |
| 无名疾病 | MPSⅧ | N-乙酰葡糖胺6-硫酸酯酶 | 成纤维细胞 | KS和HS(±)出现于尿中 | 同上 |
注:MPS-粘多糖沉积症DS-磷酸皮肤素
HS-硫酸乙酰肝素CD-硫酸软骨素
KS-硫酸角质素
遗传性粘多糖沉积症约占出生婴儿的1/30000。
(王继红 康格非)