第二节 常用基因诊断技术
当细胞的基因组DNA用特定的内切酶如Eco RⅠ切割时,凡有GAATTC的地方都被切开,得到许多长度一定但互不相等的片段,需要分析、分离的基因或DNA片段就在其中某一特定的的片段上。
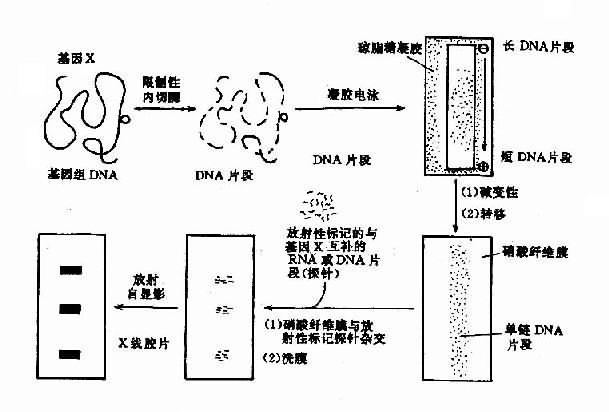
然而许多长短不同的DNA片段混合在一起是很难分析的。因此首先必需将它们按大小(长短)分离开来,这可借助凝胶电泳来完成。在电泳时,分子量愈小的片段的迁移愈快,愈大的片段愈慢。因此,在电泳结束时可以获得一个由大到小连续的带谱(smear),而由许多细胞基因组得来的某一特定片段,因其长度相同将处于同一位置,有利于检出。但凝胶易碎且操作不便。英国科学家Southern首创印迹法克服了上述困难(图13-6)。
一、Southern印迹法(Southern blot)
基本原理是:硝酸纤维膜或尼龙滤膜对单链DNA的吸附能力很强,当电泳后凝胶经过DNA变性处理,覆以上述滤膜,再于其上方压上多层干燥的吸水纸,借助它对深盐溶液的上吸作用,凝胶上的单链DNA将转移到滤膜上。转移是原位的,即DNA片段的位置保持不变。转移结束后,经过80℃烘烤的DNA,将原位地固定于膜上。
图13-6 Southern印迹杂交示意图
当含有特定基因片段已原位转移到膜上后,即可与同位素标记了的探针进行杂交,并将杂交的信号显示出来。杂交通常在塑料袋中进行,袋内放置上述杂交滤膜,加入含有变性后探针的杂交溶液后,在一定温度下让单链探针DNA与固定于膜上的单链基因DNA分子按碱基到互补原理充分结合。结合是特异的,例如只有β珠蛋白基因DNA才能结合上β珠蛋白的探针。杂交后,洗去膜上的未组合的探针,将Ⅹ线胶片覆于膜上,在暗盒中日光进行放射自显影。结合了同位素标记探针的DNA片段所在部位将显示黑色的杂交带,基因的缺失或突变则可能导致带的缺失或位置改变。
分子杂交是基因探测的基础,除了用印迹杂交外,还有斑点杂交法。即将DNA样品变性后直接点在硝酸纤维滤膜上,再与探针杂交,或者将细胞或病毒点在膜上,菌落或菌斑原位地吸附在膜上,经过变性处理,再进行杂交。斑点杂交多用于病原体基因,如微生物的基因,但也可用于检查人类基因组中的DNA序列。
二、聚合酶链反应
近年来,基因分析和基因工程技术有了革命性的突破,这主要归功于聚合酶链反应(polymerase chain reaction,PCR)的发展和应用。应用PCR技术可以使特定的基因或DNA片段在短短的2-3小时内体外扩增数十万至百万倍。扩增的片段可以直接通过电泳观察,也可用于进一步的分析。这样,少量的单拷贝基因不需通过同位素提高其敏感性来观察,而通过扩增至百万倍后直接观察到,而且原先需要一、二周才能作出的诊断可以缩短至数小时。PCR反应的原理如图13-7。
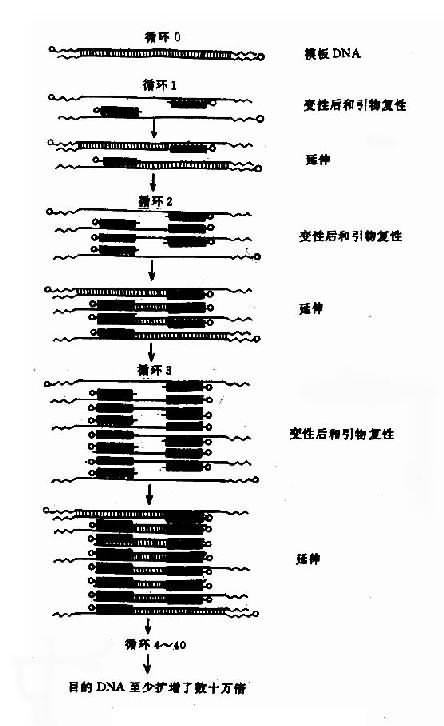
图13-7 PCR原理示意图黑色线代表引物
由图可见,首先应按照欲检测的DNA的5’和3’端的碱基顺序各合成一段长约17-20余个碱基的寡核苷酸作为引物(primer),其次是将待检测的DNA变性后,加入四种单核苷酸(dNTP)、引物和耐热聚合酶。在较低的温度,引物将与待扩增的DNA链复性结合,然后的聚合酶的作用下,利用溶液中的核苷酸原料,不断延伸合成新互补链,这样,一条DNA双链就变成了两条双链。若继续按照变性(92-95℃)→复性(40-60℃)→引物延伸(65-72℃)的顺序循环20至40个周期,就可以得到大量的DNA片段。理论上循环20周期可使DNA扩增2n,即100余万倍。PCR反应特异性强,灵敏度高,极微量的DNA即可作为扩增的模板得到大量的扩增片段。毛发、血痕,甚至单个细胞的DNA即可供PCR扩增之用。因此它用于病原体DNA的检查、肿瘤残留细胞的检出、罪犯或个体遗传物质的鉴定以及遗传病的基因诊断等。
目前已可对一系列的遗传病进行PCR诊断。如果疾病是由基因缺失引起的(如α地贫),则在缺失两端设计一对引物进行扩增,就不会得到扩增产物或只能得到缩短了的扩增产物。如果疾病是由点突变引起的,而突变的位置和性质已知,则在设计引物时使之包括突变部位,由于突变后的碱基不配对,结果无扩增片段;或者在引物设计时于其3’端设计一个错误的核苷酸,使之与突变了的核苷酸配对,其结果是正常引物不能扩增,而用错误的引物能扩增,从而可对突变的存在作出判断。
PCR技术目前有许多新的发展,用途日益扩大。例如,可用RNA为模板经过逆转录再行扩增的RT-PCR;改变两引物浓度,使其相差100倍,结果得到大量单链产物,称为不对称PCR,其单链产物可用于序列分析;在一个反应中加入多对引物同时检测多个部位的多重PCR等等。
三、扩增片段长度多态性
小卫星DNA和微卫星DNA的长度多态性可以通过PCR扩增后电泳来检出,并用于致病基因的连锁分析,这种诊断方法称为扩增片段长度多态性(amplified fragment length polymorphism,Amp-FLP)连锁分析法。PCR扩增后,产物即等位片段之间的差别有时只有几个核苷酸,故需用聚丙烯酰胺凝胶电泳分离鉴定。此法多用于突变性质不明的连锁分析.
四、等位基因的特异寡核苷酸探针诊断法
当基因的突变部位和性质已完全明了时,可以合成等基因特异的寡核苷酸探针(allele-specific oligonucleotide,ASO)用同位素或非同位素标记进行诊断。探针通常为长20bp左右的核苷酸。用于探测点突变时一般需要合成两种探针,一种与正常基因序列完全一致,能与之稳定地杂交,但不能与突变基因序列杂交;另一种与突变基因序列一致,能与突变基因序列稳定杂交,但不能与正常基因序列稳定杂交,这样,就可以把只有一个碱基发生了突变的基因区别开来.
PCR可结合ASO,即PCR-ASO技术,即先将含有突变点的基因有关片段进行体外扩增,然后再与ASO探针作点杂交,这样大大简化了方法,节约了时间,而且只要极少量的基因组DNA就可进行。
五、单链构象多态性诊断法
单链构象多态性(signle strand conformation polymorphism,SSCP)是指单链DNA由于碱基序列的不同可引起构象差异,这种差异将造成相同或相近长度的单链DNA电泳迁移率不同,从而可用于DNA中单个碱基的替代、微小的缺失或手稿的检测。用SSCP法检查基因突变时,通常在疑有突变的DNA片段附近设计一对引物进行PCR扩增,然后将扩增物用甲酰胺等变性,并在聚丙烯酰胺凝胶中电泳,突变所引起的DNA构象差异将表现为电泳带位置的差异,从而可据之作出诊断。
PCR-SSCP法具有能快速、灵敏地检测有无点突变或多态性的优点,但如欲阐明突变的碱基性质,则需作序列分析。