
第四十二章 人工合成抗菌药
第一节 喹诺酮类药物
喹诺酮类quinolones是人工合成的含4-喹诺酮基本结构,对细菌DNA螺旋酶(DNA gyrase)具有选择性抑制作用的抗菌药物。目前发展迅速,临床广为使用。
一、喹诺酮类药物概述
(一)简史
萘啶酸(nalidixic acid)是1962年用于临床的第一个喹诺酮类药(实是萘啶酮),抗菌谱窄,口服吸收差,副作用多,现已不用。吡哌酸(pipemidic acid)抗菌活性强于萘啶酸,口服少量吸收,不良反应较萘啶酸少,可用于敏感菌的尿路感染与肠道感染。1979年合成诺氟沙星(norfloxacin),随又合成一系列含氟的新喹诺酮类药,通称为氟喹诺酮类(fluoroquinolones)。
(二)化学结构与作用关系
本类药物的构效关系研究表明:4-喹诺酮母核的3位均有羧酸基,6位引入氟原子可增强抗菌作用并对金葡菌有抗菌活性;7位引进哌嗪环可提高对金葡菌及绿脓杆菌的抗菌作用(如诺氟沙星),哌嗪环被甲基哌嗪环取代(如培氟沙星),则脂溶性增加,肠道吸收增强,细胞的穿透性提高,半衰期延长。在8位引进第二个氟原子,可进一步提高肠道吸收,延长半衰期(如洛美沙星等),N-1修饰以环丙基团(环丙沙星)或噁嗪基团(氧氟沙星)可扩大抗菌谱,增强对衣原体、支原体及分支杆菌(结核杆菌与麻风杆菌等)的抗菌活性,噁嗪环还可提高水溶性,使药物在体内不被代谢,以原形经尿排泄。

(三)抗菌作用机制
喹诺酮类通过抑制DNA螺旋酶作用,阻碍DNA合成而导致细菌死亡。大肠杆菌的DNA螺旋酶是四叠体结构的蛋白,由2个A亚单位与2个B亚单位组成,分子量分别为105kD与95kD(见图42-1)。细菌在合成DNA过程中,DNA螺旋酶的A亚单位将染色体DNA正超螺旋的一条单链(后链)切开,接着B亚单位使DNA的前链后移,A亚单位再将切口封住,形成了负超螺旋。根据实验研究,氟喹诺酮类药并不是直接与DNA螺旋酶结合,而是与DNA双链中非配对碱基结合,抑制DNA抑螺旋酶的A亚单位,使DNA超螺旋结构不能封口,这样DNA单链暴露,导致mRNA与蛋白合成失控,最后细菌死亡。
本类药体外对DNA螺旋酶的半抑制浓度(IC50)与其对细菌的MIC呈一定的平行关系。哺乳动物的细胞内也含有生物活性与细菌DNA螺旋酶相似的酶,称为拓朴异构酶Ⅱ(topoisomerase Ⅱ)。氟喹诺酮类药对人体细胞拓朴异构酶Ⅱ影响较小(见表42-1)。从该表可见氧氟沙星与环丙沙星对动物细胞内拓朴异构酶Ⅱ的作用明显比依诺沙星与萘啶酸小,IC50很高,选择指数很大。这可能是氧氟沙星与环丙沙星不良反应较少的原因。
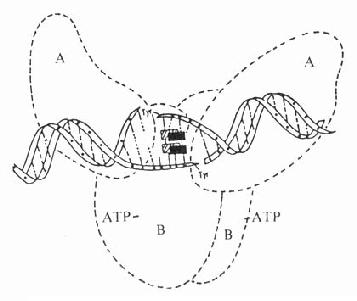
图42-1 喹诺酮类药物-DNA结合抑制DNA螺旋酶活性的示意图
图中实心和斜线长方形示喹诺酮类药物分子,A、B为DNA螺旋酶的A、B亚单位。在DNA螺旋酶作用下,DNA双链打开,而药物分子嵌入双链。与非配对碱基结合,阻碍DNA双链封口
(四)细菌耐药机制
氟喹诺酮类药物广泛应用后,已出现细菌耐药性。耐药机理研究证实主要是染色体突变,不存在质粒介导的耐药性。耐药机制有二:①细菌DNA螺旋酶的改变,与细菌高浓度耐药有关;②细菌细胞膜孔蛋白通道的改变或缺失与低浓度耐药有关。耐药菌株DNA螺旋酶的活性改变主要由于gyrA基因突变所致。
(五)氟喹诺酮类药理学共同特性
①抗菌谱广,尤其对革兰阴性杆菌包括绿脓杆菌在内有强大的杀菌作用,对金葡菌及产酶金葡菌也有良好抗菌作用;某些品种对结核杆菌,支原体,衣原体及厌氧菌也有作用;②细菌对本类药与其他抗菌药物间无交叉耐药性;③口服吸收良好,部分品种可静脉给药;体内分布广,组织体液浓度高,可达有效抑菌或杀菌水平;血浆半衰期相对较长,大多为3~7小时以上。血浆蛋白结合率低(14%~30%),多数经尿排泄,尿中浓度高;④适用于敏感病原菌所致的呼吸道感染、尿路感染、前列腺炎,淋病及革兰阴性杆菌所致各种感染,骨、关节、皮肤软组织感染;⑤不良反应少(5%~10%),大多轻微,常见的有恶心、呕吐、食欲减退、皮疹、头痛、眩晕。偶有抽搐精神症状,停药可消退。
表42-1 喹诺酮类药物对大肠杆菌和哺乳动物细胞DNA旋转酶的选择作用
| 药物 | IC50(mg/L) | 选择指数 B/A |
|
| A 大肠杆菌KL-16DNA螺旋酶 |
B 胎牛胸腺局部拓朴异构酶Ⅱ |
||
| 氧氟沙星 | 0.76 | 1870 | 2461 |
| 环丙沙星 | 0.13 | 155 | 1192 |
| 依诺沙星 | 1.72 | 93 | 54 |
| 萘啶酸 | 23.0 | 385 | 17 |
二、各种喹诺酮类药特点
吡哌酸(pipemidic acid,PPA)对革兰阴性菌的抗菌作用较萘啶酸强,对革兰阳性和部分绿脓杆菌有一定作用。口服400mg后血浓度达不到治疗效果,但尿中浓度高,可达900mg/L以上,主要用于治疗尿路和肠道感染。
诺氟沙星(norfloxacin)又名氟哌酸,是第一个氟喹诺酮类药,抗菌谱广,抗菌作用强,对革兰阳性和阴性菌包括绿脓杆菌均有良好抗菌活性,明显优于吡哌酸。口服吸收约35%~45%;易受食物影响,空腹比饭后服药的血浓度高2~3倍,血浆蛋白结合率为14%,体内分布广,组织浓度高,药物消除半衰期为3~4小时。主要用于尿路及肠道感染。
氧氟沙星(ofloxacin)又名氟嗪酸,抗菌活性强,对革兰阳性菌(包括甲氧西林耐药金葡菌,MRSA)革兰阴性菌包括绿脓杆菌均有较强作用;对肺炎支原体,奈瑟菌病,厌氧菌及结核杆菌也有一定活性。对感染小鼠的保护效果明显强于诺氟沙星、依诺沙星。口服吸收快而完全,血药浓度高而持久,血浆消除半衰期为5~7小时,药物体内分布广,尤以痰中浓度较高,70%~90%药物经肾排泄,48小时尿中药物浓度仍可达到对敏感菌的杀菌水平,胆汁中药物浓度约为血药浓度的7倍左右。
依诺沙星(enoxacin)又名氟啶酸,抗菌谱和抗菌活性和诺氟沙星相似,对厌氧菌作用较差。口服吸收好,不受食物影响,血药浓度介于诺氟沙星与氧氟沙星之间,口服后约50%~65%经肾排泄,消除半衰期为3.3~5.8小时。副作用以消化道反应为主,偶有中枢神经系统毒性反应。
培氟沙星(pefloxacin)又名甲氟哌酸,抗菌谱广与诺氟沙星相似,抗菌活性略逊于诺氟沙星,对军团菌及MRSA有效,对绿脓杆菌的作用不及环丙沙星。口服吸收好,生物利用度为90%~100%。血药浓度高而持久,半衰期可达10小时以上,体内分布广泛,尚可通过炎症脑膜进入脑脊液。
环丙沙星(ciprofloxacin)又名环丙氟哌酸,抗菌谱广,体外抗菌活性为目前在临床应用喹诺酮类中最强,对耐药绿脓杆菌,MRSA,产青霉素酶淋球菌、产酶流感杆菌等均有良效,对肺炎军团菌及弯曲菌亦有效,一些对氨基甙类、第三代头孢菌素等耐药的革兰阴性和阳性菌对本品仍然敏感。口服后本品生物利用度为38%~60%,血浓度较低,静脉滴注可弥补此缺点。半衰期为3.3~5.8小时,药物吸收后体内分布广泛。
洛美沙星(lomefloxacin)抗菌谱广,体外抗菌作用与诺氟沙星、氧氟沙星、氟罗沙星相似,但比环丙沙星弱;体内抗菌活性比诺氟沙星与氧氟沙星强,但不及氟罗沙星。本品口服吸收好,生物利用度为85%,血药浓度高而持久,半衰期约7小时,体内分布广,药物经肾排泄。
氟罗沙星(fleroxacin)又名多氟沙星,抗菌谱广,体外抗菌活性略逊于环丙沙星,但其体内抗菌活性强于现有各喹诺酮药。口服吸收好,生物利用度达99%。口服同剂量(400mg)的血药浓度比环丙沙星高2~3倍,半衰期为9小时。体内分布广,药物经肾排泄,约为给药量50%~60%。
表42-2 几种常用氟喹诺酮类药的药代动力学参数
药物 |
单次口服剂量(mg) |
Cmax(mg/L) |
t1/2kel(h) |
绝对生物利用度(%) |
Vd(L) |
总清除率(L/h) |
原药尿中排泄率(%) |
粪便排泄率 |
| 诺氟沙星 | 400 | 1.58 | 3~4 | 35~45 | >100 | 51.6 | 25~30 | 28 |
| 培氟沙星 | 400 | 3.80 | 7.5~11 | 90~100 | 139 | 8.94 | 11 | |
| 依诺沙星 | 400 | 3.70 | 3.3~5.8 | 80~89 | 175 | 21.0 | 52 | 18 |
| 氧氟沙星 | 400 | 5.60 | 5.0~7.0 | 85~95 | 120 | 12.84 | 70~80 | 4 |
| 环丙沙星 | 500 | 2.56 | 3.3~4.9 | 38~60 | 307 | 39.12 | 29~44 | 15 |
三、应用注意事项
1.对幼年动物可引起软骨组织损害,故不宜用于妊娠期妇女和骨骼系统未发育完全的小儿。药物可分泌于乳汁,乳妇应用时应停止哺乳。
2.可引起中枢神经系统不良反应,不宜用于有中枢神经系统病史者,尤其是有癫痫病史的患者。
3.可抑制茶碱类、咖啡因和口服抗凝血药在肝中代谢,使上述药物浓度升高引起不良反应。产生上述相互作用最显著者为依诺沙星、其次为环丙沙星与培氟沙星,氧氟沙星无明显影响。因此应避免与有相互作用的药物合用,如有指征合用时,应对有关药物进行必要的监测。
4.制酸药的同时应用,可形成络合物而减少其自肠道吸收,宜避免合用。
5.肾功能减退者应用主要经肾排的药物如氧氟沙星和依诺沙星应减量。